



作者:中国医学科学院北京协和医院神经外科刘子源、冯铭、王任直
郎格汉斯组织细胞增多症(Langerhans cell histiocytosis,LCH),又称组织细胞增多症X(histiocytosisX),是一组以表型类似于表皮郎格汉斯细胞的树突状细胞增生为共同特征的异质性疾病,常累及多脏器,如骨骼、皮肤、中枢神经系统、肺、肝、脾、淋巴结等,诊断高度依赖组织病理。LCH中枢神经系统受累以下丘脑-垂体病变最多见。由于LCH为少见病,单纯累及下丘脑-垂体而无其他系统受累表现的LCH常常被误诊为其他鞍区疾病。现报道本院收治的1例孤立性鞍区LCH,并简要讨论LCH尤其是累及下丘脑-垂体的LCH的诊断、治疗要点;以加深临床医生对LCH的认识。

临床资料
患者:男性,14岁,1月前无明显诱因出现烦渴、多饮、多尿,尿量与饮水量大致相当,尿色清亮。当地医院查尿比重1.005,余(-);禁水-加压试验:禁水8小时,尿量110-330ml/h,尿比重1.003-1.006,予5U垂体后叶素后尿量减少至15-60ml/h,尿比重1.010-1.018;考虑“中枢性尿崩症”收入我院。
完善相关检查:内分泌方面,GH、IGF1、FSH、LH、ACTH、F、24hUFC、TSH、FT3、FT4水平均正常,血PRL18.14ng/ml↑ (2.64-13.13);血AFP、CEA、β-HCG水平均正常;血ACE水平正常;脑脊液常规、生化均(-)。
影像学方面:垂体平扫+增强MRI提示“垂体柄增粗,均匀明显强化,垂体后叶T1高信号消失”(图1);胸HRCT+增强提示“右肺下叶小结节”;甲状腺超声提示“甲状腺弥漫性病变”;全身骨显像未见异常。
初步诊断考虑“鞍区生殖细胞瘤”,计划行活检明确诊断。患者于我院行经鼻蝶窦入路鞍区探查术,术中在鞍内可见质韧、灰白色的肿瘤组织,血供不丰富;组织采取钳留取肿瘤标本组织送检,冰冻病理回报“少许肿瘤组织伴大量嗜酸细胞浸润,不除外郎格汉斯组织细胞增多症”;石蜡病理回报“病变符合郎格汉斯组织细胞增多症”(图2),免疫组化AE1/AE3(+) ,CD1a(+),Langerin(+),CD34(+) ,CD68(+),CgA(+),EMA(-),Ki-67(index1%),OCT3/4(-),S-100(+) ,SALL-4(-),Vimentin(+),GFAP(-) ,Calretinin(-),WT-1(-)。确诊为“郎格汉斯组织细胞增多症”。
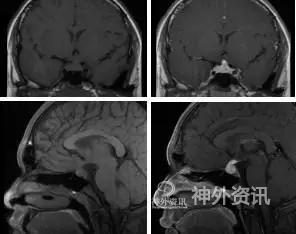
图1. 垂体增强MRI A&B. 冠状位,可见垂体柄增粗,均匀明显强化;C&D. 矢状位,可见垂体后叶T1高信号消失。
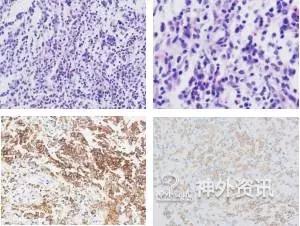
图2.组织病理结果 。A. HE染色(×400);B. HE染色(×1000),可见核沟(箭头所示);C. CD1a免疫组化(×400),可见弥漫CD1a(+)细胞;D. Langerin免疫组化(×400),可见散在Langerin(+)细胞。
讨论
郎格汉斯组织细胞增多症(Langerhans cell histiocytosis,LCH)是一种罕见疾病,主要发生于婴幼儿、儿童及青少年,成年人LCH仅占30%[1]。一项针对0-19岁人群的队列研究[2]表明,LCH发病率约百万分之0.7,男女比例大致相当,居住环境拥挤(RR=1.84)、教育水平较低(RR=1.49)均为LCH发生的危险因素。LCH可累及多脏器,其中以骨骼(80%)、皮肤(25%)、下丘脑-垂体(25%)最为常见,其他容易受累的脏器还有肺、肝、脾、淋巴结等。
根据受累系统的多少,可将LCH分为单系统受累LCH(single systemLCH,SS-LCH)及多系统受累LCH(multi systemLCH,MS-LCH)两大类。其中,肝、脾、造血系统、肺受累称为风险器官受累(risk organ involve ment)。MS-LCH[3,4,5]、颅面部受累(cranio facial LCH)[6]的患者更容易出现下丘脑-垂体受累。LCH累及下丘脑-垂体时,取决于具体受累部位不同,可表现为中枢性尿崩症、垂体前叶功能减退、视野缺损等[7]。本例中,患者直接以中枢性尿崩症起病。
孤立性累及下丘脑-垂体的LCH非常少见。Prosch等[8]在就诊时即已查出下丘脑-垂体外病变,另外51%在初次就诊1年内出现下丘脑-垂体外病变;最终累计仅5例(9%)单纯累及下丘脑-垂体,其余49例(91%)均出现下丘脑-垂体外病变。可见,对于以中枢性尿崩症起病、拟诊LCH的患者,除了努力取得组织病理证据外,寻找支持LCH诊断的颅外病变证据也非常重要。
影像学上,累及下丘脑-垂体的LCH最常表现为MRI上T1序列低-等信号、T2序列等-高信号,均匀强化,最常累及垂体柄,表现为垂体柄增粗。Groi s等人[9]总结了59例中枢性尿崩症患者影像学表现,表明垂体柄增粗常出现于尿崩症之前,其与尿崩症严重程度及对治疗的反应均不平行;提示垂体柄增粗与中枢性尿崩症可能并无严格对应关系。此外,T1序列上垂体后叶高信号(表征正常神经垂体)常常消失,提示垂体后叶加压素分子的减少、缺失,与尿崩症严重程度及对治疗反应相平行[10]。本例中,患者垂体平扫+增强MRI提示“垂体柄增粗,均匀明显强化,垂体后叶T1高信号消失”,与上均相符。
影像学表现为垂体柄增粗的患者在临床中可能存在不同的诊治策略,如随访观察、立体定向活检等。Leger等人[11]随访了26例表现为垂体柄增粗及中枢性尿崩症的儿童患者,其中5例最终诊断为LCH,4例诊断为生殖细胞瘤,17例诊断为特发性中枢性尿崩症;表明中枢性尿崩症、垂体柄增粗不是LCH的特异表现。曾有观点认为垂体柄增粗结合支持LCH的颅外病变可临床诊断为LCH;但Prosch等人[12]报道1例表现为垂体柄增粗、颈椎椎体病变的患者,按LCH试验性化疗后病灶增大,最终经手术活检确认为生殖细胞瘤;表明LCH的临床诊断仍然不特异,仅病理诊断具有确诊意义。然而考虑到垂体柄活检本身存在风险,需在获益-风险间作一权衡。Grois等人[13]总结得出,对于孤立性下丘脑-垂体受累或下丘脑-垂体病灶直径>6mm的病灶,需要行活检以明确诊断。Robinson等[14]回顾分析了69例影像学表现为垂体柄增粗的儿童患者,表明伴有中枢性尿崩症、垂体前叶功能减退或随访提示垂体柄进行性增大的患者更可能为肿瘤性疾病(如生殖细胞瘤、颅咽管瘤),这些患者可能更需要积极活检。
下丘脑-垂体部位活检可存在多种术式。Jian F等人[15]报道了17例影像学表现为垂体柄增粗的中枢性尿崩症患者经眶上锁孔入路开颅活检的病例,其中6例诊断为LCH,10例为生殖细胞瘤,1例为Erdheim-Chester病。Jinguji S[16]等人报道了11例影像学表现为垂体柄增粗并于神经内镜下行垂体柄活检的病例,均明确了诊断,且术后均未出现进一步的垂体前叶功能减退等并发症。这提示在对垂体柄增粗患者进行充分的随访及评估后,可进行垂体柄活检以帮助鉴别诊断。
根据国际组织细胞学会2009年发布的LCH诊断标准[17],病灶组织普通HE染色下查得“咖啡豆样”细胞核、核沟等征象,可作为提示LCH的证据。免疫组化满足CD1a(+)和/或Langerin(+),结合临床表现,可建立LCH的诊断。本例中,病理活检光镜下可见核沟等典型结构,免疫组化提示CD1a(+)、Langerin(+),故确诊LCH。
除累及下丘脑-垂体的LCH外,多种病变在影像学上均可表现为垂体柄增粗,故而LCH以下丘脑-垂体受累为首发及唯一表现时很容易造成误诊,因而鉴别诊断具有重要意义。现就2个发病率较高的且容易与LCH相混淆的疾病鉴别诊断简要讨论如下:
生殖细胞肿瘤(germ cell tumors,GCTs),其中又可细分为生殖细胞瘤(germinomas)及非生殖细胞来源的生殖细胞肿瘤(NGGCTs)。好发于儿童及青少年,最常见于松果体区及鞍上区。其影像学多表现为T1序列低-中信号、T2序列中-高信号,均匀强化,与LCH难以鉴别。但由于GCTs容易沿脑脊液播散,脑脊液细胞学查得瘤细胞及脊髓MRI检查发现柔脑脊膜播散更支持GCTs的诊断。另一方面,血清及脑脊液中β-HCG、AFP水平升高更支持GCTs尤其是NGGCTs的诊断。然而,β-HCG、AFP作为肿瘤标志物既不灵敏也不特异。如绝大多数生殖细胞瘤并不伴有血清、脑脊液β-HCG、AFP的升高;并且血清、脑脊液β-HCG、AFP水平升高也并不特异;如Kinoshita等[18]曾报道1例脑脊液β-HCG水平升高的LCH。病理方面,PLAP是GCTs特异的免疫组化标志物,可资鉴别。
经垂体炎(lymphocytic infundibuloneuro hypophysitis, LINH)。即淋巴细胞性垂体炎(lymphocytic hypop hysitis)累及神经垂体的亚型。常见于女性尤其是妊娠后期或产后的年轻女性[19]。影像学为T1等信号、T2中-高信号,均匀强化,与LCH难以鉴别。但值得注意的是孤立性LINH少见,常可合并腺垂体受累或其他自身免疫性疾病[20],且LINH存在自限性,常自行缓解,可资鉴别。
LCH具体治疗方案应根据病情制定;部分LCH具有自发缓解倾向[21,22]。根据国际组织细胞学会2009年发布的LCH治疗指南,MS-LCH,或SS-LCH伴颅面部受累、多发性骨损害或病灶位于重要解剖位置的,均需要接受系统性治疗。首选治疗方案为长春碱联合泼尼松化疗,并于6周后进行再评估,确定是否继续治疗方案或改用补救治疗方案。
参考文献
1. Malpas J S. Langerhans cell histiocytosis in adults. Hematol Oncol Clin North Am, 1998, 12(2): 259-268.
2.Ribeiro K B, Degar B, Antoneli C B, et al. Ethnicity, race, and socioeconomic status influence incidence of Langerhans cell histiocytosis. Pediatr Blood Cancer, 2015, 62(6): 982-987.
3. Lin K D, Lin J D, Hsu H H, et al. Endocrinological aspects of Langerhans cell histiocytosis complicated with diabetes insipidus. J Endocrinol Invest, 1998,21(7): 428-433.
4. .Nanduri V R, Bareille P, Pritchard J, et al. Growth and endocrine disorders in multisystem Langerhans' cell histiocytosis. Clin Endocrinol (Oxf), 2000, 53(4): 509-515.
5. .Grois N, Flucher-Wolfram B, Heitger A, et al. Diabetes insipidus in Langerhans cell histiocytosis: results from the DAL-HX 83 study. Med Pediatr Oncol, 1995, 24(4): 248-256.
6. .Grois N, Potschger U, Prosch H, et al. Risk factors for diabetes insipidus in Langerhans cell histiocytosis. Pediatr Blood Cancer, 2006, 46(2): 228-233.
7. .Dunger D B, Broadbent V, Yeoman E, et al. The frequency and natural history of diabetes insipidus in children with Langerhans-cell histiocytosis. N Engl J Med, 1989, 321(17): 1157-1162.
8. .Prosch H, Grois N, Prayer D, et al. Central diabetes insipidus as presenting symptom of Langerhans cell histiocytosis. Pediatr Blood Cancer, 2004, 43(5): 594-599.
9. Grois N, Prayer D, Prosch H, et al. Course and clinical impact of magnetic resonance imaging findings in diabetes insipidus associated with Langerhans cell histiocytosis. Pediatr Blood Cancer, 2004, 43(1): 59-65.
10. Chaudhary V, Bano S, Aggarwal R, et al. Neuroimaging of Langerhans cell histiocytosis: a radiological review. Jpn J Radiol, 2013, 31(12): 786-796.
11. Leger J, Velasquez A, Garel C, et al. Thickened pituitary stalk on magnetic resonance imaging in children with central diabetes insipidus. J Clin Endocrinol Metab, 1999, 84(6): 1954-1960.
12. Prosch H, Grois N, Bokkerink J, et al. Central diabetes insipidus: Is it Langerhans cell histiocytosis of the pituitary stalk A diagnostic pitfall. Pediatr Blood Cancer, 2006, 46(3): 363-366.
13. Grois N, Fahrner B, Arceci R J, et al. Central nervous system disease in Langerhans cell histiocytosis. J Pediatr, 2010, 156(6): 873-881, 881.
14. Robison N J, Prabhu S P, Sun P, et al. Predictors of neoplastic disease in children with isolated pituitary stalk thickening. Pediatr Blood Cancer, 2013, 60(10): 1630-1635.
15. Jian F, Bian L, Sun S, et al. Surgical biopsies in patients with central diabetes insipidus and thickened pituitary stalks. Endocrine, 2014, 47(1): 325-335.
16. .Jinguji S, Nishiyama K, Yoshimura J, et al. Endoscopic biopsies of lesions associated with a thickened pituitary stalk. Acta Neurochir (Wien), 2013, 155(1): 119-124, 124.
17. Histocyte Society. Langerhans Cell Histiocytosis: Evaluation and Treatment Guidelines. 2009.
18. Kinoshita Y, Yamasaki F, Usui S, et al. Solitary Langerhans cell histiocytosis located in the neurohypophysis with a positive titer HCG-beta in the cerebrospinal fluid. Childs Nerv Syst, 2015.
19. Beressi N, Beressi J P, Cohen R, et al. Lymphocytic hypophysitis. A review of 145 cases. Ann Med Interne (Paris), 1999, 150(4): 327-341.
20. Crock P. Lymphocytic hypophysitis. Curr Opin Endocrinol Diabetes, 1997, 4: 115-123.
21. Fahrner B, Prosch H, Minkov M, et al. Long-term outcome of hypothalamic pituitary tumors in Langerhans cell histiocytosis. Pediatr Blood Cancer, 2012, 58(4): 606-610.
22. Nagasaki K, Tsumanuma I, Yoneoka Y, et al. Spontaneous regression of isolated neurohypophyseal langerhans cell histiocytosis with diabetes insipidus. Endocr J, 2009, 56(5): 721-725.
