


本文源自公众号:神经病学思辨
目前学术界较一致的看法是,导致帕金森病(PD)患者的核心病理特征是黑质的多巴胺能神经元退化、凋亡,导致纹状体多巴胺水平下降,引发运动障碍。但是人们对造成神经细胞凋亡或退行性变的分子机制并不完全了解。故而当前对帕金森病的治疗只是治标,即现有的治疗方法并不能减缓黑质多巴胺神经凋亡或退行性变。
在病理学研究方面,原发性帕金森病脑干切面可见中脑黑质、脑桥蓝斑存在不同程度的色素细胞缺失。光学显微镜下可见黑质外侧带色素细胞萎缩、变性和大量缺失,伴神经胶质细胞增生,在残留神经元内发现特征性路易小体。路易小体被认为是原发性帕金森病的组织病理学标志物。是一种神经元胞质包涵体,包括两种类型,即经典型和皮质型。经典型路易小体又称脑干型,主要见于黑质、蓝斑、无名质、迷走神经背核、中缝核、脊髓中间内外侧柱、大脑脚脑桥核、E—w核,亦可见于周围交感和副交感神经元。另外,脑干神经元内还可见苍白小体(pale body),是一种具有较多颗粒样成分的嗜伊红包涵体,缺乏周晕,可能是路易小体的前体结构。
病理研究还发现除黑质一纹状体系统变性外,原发性帕金森病也存在其他结构的组织病理学改变:脑干中缝核、脑桥蓝斑、延髓迷走神经背核、外周交感神经节,以及心脏和胃肠自主神经丛均存在不同程度的神经元缺失和路易小体、路易轴索变性。黑质.纹状体系统外的结构性病变与帕金森病的非运动症状存在密切关联性。
目前对路易小体形成的机制尚不十分清楚。采用各种抗体标记的免疫组织化学染色发现路易小体共含有20余种蛋白质成分,主要为突触共核蛋白、神经微丝蛋白(NF)和泛素蛋
白(ubiquitin)。细胞内泛素.蛋白酶体系统(UPS)对路易小体中丝状蛋白沉积所致细胞反应起重要作用,因此,路易小体内可检测到泛素蛋白、.晶状体蛋白(B—crystallin)等与蛋白质降解相关的成分。
路易小体内泛素蛋白的存在有两种解释,一种可能是,丝状蛋白沉积所诱发的依赖泛素蛋白的蛋白质降解通路病理性反应;另一种可能是,由于某种底物蛋白或降解系统中蛋白酶异常而产生有害蛋白质分解。有关研究进一步发现,α突触核蛋白首先在黑质,特别在神经元的线粒体聚积,此时,在黑质中发现有不少多巴胺神经元凋亡。
自1998年发现.突触共核蛋白是路易小体较特异性的蛋白抗原成分以来,抗d一突触共核蛋白抗体免疫组织化学染色已逐渐成为路易小体相关疾病的主要病理诊断方法,同时,突触共核蛋白的发现也促进了对路易小体相关疾病发病机制的进一步认识。帕金森病的神经退行性变与遗传因素、神经细胞凋亡的多样性密切相关,主要涉及多巴胺能神经元的退化、α-突触核蛋白异常聚集及多种细胞死亡机制。
人们就帕金森病的细胞凋亡发病机制提出种种假说,比如多巴胺缺失、内源性MPTP毒素、钙失衡、氧化自由基、线粒体功能失调及炎症反应等。因此有学者提出神经元在帕金森病患者脑中发生退行性变可能有三种通路:
第一,死亡受体及凋亡因子可通过线粒体而地导致多巴胺神经元凋亡;
第二,超氧化物歧化酶(SOD)可以增加氧自由基
第三,蛋白在高尔基体内调节皮质神经元凋亡。
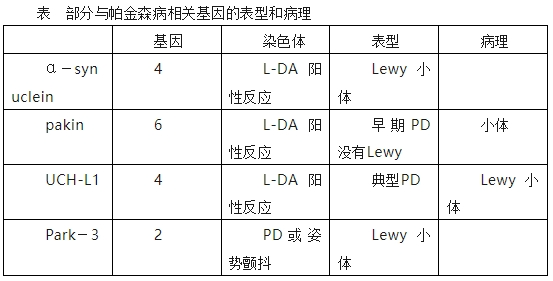
约5-10%的帕金森病病例与遗传突变相关,如GBA 、SNCA、LRRK2、PARK2、 PRKN、PINK1、 PARK7、NCA、VPS35等基因突变可能导致蛋白质错误折叠、线粒体功能障碍或异常蛋白聚集,进而引发神经元损伤。早发型帕金森病(20-40岁发病)中,约90%与遗传因素相关。在极少数情况下,帕金森病可能不产生路易体。例如,由于Parkin突变。
近年遗传学方面有较多的进展,迄今为止,已发现与帕金森病有关的多种突变基因。以α突触核蛋白(α-synuclein)为例,最近有研究发现,在α突触核蛋白转基因鼠的黑质中有不少多巴胺神经细胞凋亡,但在皮层却没有发现许多神经细胞凋亡。这种神经细胞凋亡或退行性变分布与帕金森病患者脑部病变的病理一致,值得注意的是,α突触核蛋白转基因鼠的脑中,皮层神经细胞以及海马神经细胞释放的谷氨酸要比黑质神经细胞多得多。因此,α突触核蛋白在不同的脑区或不同的神经元可能有不同的功能。治疗帕金森病的一大挑战在于无法预测该病在不同患者身上的表现。经常是PD患者之间临床治疗的经验值完全不同。(如症状、发病年龄、进展速度甚至治疗反应)。遗传学可能有助于解释其中一些差异。
死亡受体说及凋亡因子方面,近年来研究发现在帕金森病患者脑中肿瘤坏死因子(TNF)在黑质中显著增加,同时研究者发现,在试验猴和小鼠动物模型这也有类似的现象。实验证明,TNF对多巴胺神经元有毒性作用。TNF如何导致多巴胺神经元死亡?
学术界通常认为,TNF有3个与死亡有关的受体。分别是1,Fas细胞表面死亡受体(Fas cell surface death receptor);2,肿瘤坏死因子受体(TNFRI);3,P75NGF低亲和力神经生长因子受体(Low Affinity Neurotrophin Receptor)。这3个死亡受体的共同特征就是在它们基因的N端(细胞内端)都有一个片断称之“死亡基因段”(death domain)。一旦这个死亡基因段被激活,便会引发一系列不同的导致动物的信号传导通路。比如:
1,激活TNFRI,会引发肿瘤坏死因子受体相关死亡结构域蛋白(TRADD:Tumor Necrosis Factor Receptor-Associated Death Domain Protein)的活性,接着激发IKKs(IKKs是NF-κB信号通路的核心激酶)和核转录因子(NFKb),最后导致动物细胞凋亡。
2,FAS则是通过激活FADD(Fas相关死亡域蛋白Fas-associated death domain protein,又称Mort1)、CASPASE-8(Caspase:含半胱氨酸的天冬氨酸蛋白水解酶(cysteinyl aspartate-specific proteinase)和核转录因子(NFkb)而引起的凋亡。
3,P75NGF是通过线粒体产生过多的自由基,释放大量细胞色素C,激活凋亡引导因子Apaf-1,最后激活CASPASE-3而引起细胞的死亡。
研究发现,在帕金森病患者的脑中有很多肿瘤坏死因子受体(TNFRI),尤其在黑质的多巴胺神经元中居多,TNFRI是TNFα引起神经元死亡的原因之一,肿瘤坏死因子(TNF)引起的神经元死亡或退行性变是通过其信号NF-kb的胞核转移完成的。
研究还发现在阿尔茨海默病和帕金森病患者脑中凋亡诱导因子(APAF-1)也有显著增加。在α突触核蛋白转基因动物凋亡的神经元中积聚了大量的凋亡诱导因子。小胶质细胞产生很多氧化多巴胺导致神经元的死亡。
上述针对帕金森病神经变性相关遗传学和神经细胞凋亡多样性研究取得一定的进展,进一步加强了学者们有关帕金森病是由基因、环境和生活方式相互作用引起的这一病因推论。帕金森病的神经退变是遗传背景与凋亡程序动态互作的结果。个体基因谱差异决定了神经元对特定凋亡通路的敏感性,而凋亡多样性则解释了临床表型与进展速率的差异。如何更好地理解这些因素相互致病的复杂作用,对于提升帕金森病的防治水平至关重要。
![]()
声明:脑医汇旗下神外资讯、神介资讯、神内资讯、脑医咨询、Ai Brain 所发表内容之知识产权为脑医汇及主办方、原作者等相关权利人所有。
