





[REF:Yupei Guo, Zian Li, Leslie A Parsels, Zhuwen Wang, Joshua D Parsels, Anushka Dalvi, Stephanie The, Nan Hu, Victoria M Valvo, Robert Doherty, Erik Peterson, Xinjun Wang, Sujatha Venkataraman, Sameer Agnihotri, Sriram Venneti, Daniel R Wahl, Michael D Green, Theodore S Lawrence, Carl Koschmann, Meredith A Morgan, Qiang Zhang, H3K27M diffuse midline glioma is homologous recombination defective and sensitized to radiotherapy and NK cell-mediated antitumor immunity by PARP inhibition, Neuro-Oncology, 2025;, noaf097, https://doi.org/10.1093/neuonc/noaf097]
摘 要
简 介
H3K27突变型弥漫中线胶质瘤(DMG H3K27-altered)是一种以组蛋白H3第27位赖氨酸突变至甲硫氨酸(H3K27M)为特征、预后极差的儿童高级别脑肿瘤。分割放疗(fractionated radiotherapy, RT)是当前标准治疗手段,可缓解症状并延缓肿瘤进展,患儿基本不可避免的还是会出现肿瘤复发。找到H3K27M突变型肿瘤细胞潜在的脆弱性(vulnerability;achilles' heel)从而与放疗相结合以达到更加有效地抑制肿瘤并提高患儿生存期具有非常重要的临床意义。
在弥漫中线胶质瘤(DMG)中,H3K27M突变是一种功能获得性突变,可通过抑制多梳抑制复合体2(PRC2)的甲基转移酶活性,引发顺式(cis)与反式(trans)双重作用的表观遗传重编辑,导致抑制性三甲基化标记(H3K27me3)的全域性丢失及激活型乙酰化修饰(H3K27ac)的增加。放疗通过诱导组蛋白翻译后修饰(如泛素化与甲基化)进一步重塑表观基因组,这些修饰对DNA修复蛋白的募集至关重要。Siddaway等研究发现H3K27M突变可改变DNA修复蛋白的结合模式,但其介导的表观遗传改变与放疗之间的特异性分子互作机制仍不明确。两者存在协同作用的证据包括:药物抑制组蛋白去甲基化酶JMJD3/UTX可通过恢复H3K27me3水平增强放疗敏感性,而抑制溴结构域蛋白BRD4则可阻断其与H3K27ac的结合进而实现放疗增敏。除H3K27ac升高外,H3K27M突变还会显著增加组蛋白H4第16位赖氨酸(H4K16ac)及组蛋白H3第9、64位赖氨酸(H3K9ac、H3K64ac)的乙酰化水平。这些改变共同提示H3K27M突变导致染色质高度乙酰化并呈现开放状态,可能削弱连接组蛋白H1与染色质的结合能力。鉴于组蛋白H1在DNA损伤应答(DDR)中的关键作用,H3K27M突变如何通过影响组蛋白H1调控放疗诱导的DNA修复,其分子机制仍有待阐明。
除直接诱导肿瘤细胞致死性DNA损伤外,放疗(RT)还可激活抗肿瘤免疫应答。放疗诱导的DNA损伤通过增强肿瘤抗原性、缓解免疫抑制状态,同时激活固有免疫与适应性免疫应答。我们团队及其他研究组已证实,DNA损伤应答(DDR)抑制剂可协同放疗增强免疫原性DNA/RNA的释放,并通过I型干扰素(T1IFN)通路强化抗肿瘤免疫效应。此外,DDR抑制剂与放疗联用能进一步在肿瘤微环境(TME)中启动并激活细胞毒性T细胞、自然杀伤(NK)细胞等抗肿瘤免疫细胞。然而,H3K27M突变型DMG特有的免疫“冷”微环境特征及有限的淋巴细胞浸润,为免疫治疗策略的研发带来严峻挑战。尽管具有免疫惰性表型,DMG肿瘤内仍存在淋巴细胞浸润——体外及体内实验表明,相较于CD8+ T细胞,NK细胞对DMG肿瘤细胞展现出更强的杀伤活性。临床证据显示,在H3K27M DMG患者中,瘤内NK细胞(而非CD8+ T细胞)的浸润水平与总生存期呈正相关,这一发现为探索增强NK细胞抗肿瘤免疫应答的治疗策略提供了重要理论依据。
自然杀伤(NK)细胞通过分泌颗粒酶、穿孔素及细胞因子直接发挥肿瘤细胞杀伤作用。其功能执行受表面激活型与抑制型受体的动态调控。其中,激活型受体NKG2D(自然杀伤细胞活化性受体2D)通过识别肿瘤细胞表面配体(人类中主要为MICA、MICB和ULBP1-6;小鼠中为Rae1、H60和Mult1)触发NK细胞脱颗粒、细胞因子释放及裂解活性。值得注意的是,放疗或基因毒性药物诱导的DNA损伤可显著上调NKG2D配体表达。因此,通过增强放疗介导的NKG2D配体呈递,可能打破DMG肿瘤细胞的免疫逃逸平衡,激活NK细胞主导的抗肿瘤免疫应答。
本研究中,我们首次发现H3K27M突变通过破坏连接组蛋白H1与高乙酰化核心组蛋白的相互作用,抑制放疗诱导的H1 Lys63(K63)位点多聚泛素化修饰,进而阻碍同源重组修复(HRR)关键蛋白(RNF168、BRCA1、RAD51)的募集,最终导致同源重组修复功能缺陷。这一发现促使我们提出假设:在H3K27M突变型弥漫中线胶质瘤(DMG)中,可利用PARP抑制剂靶向HRR缺陷,实现放疗与增强放疗诱导免疫原性的双重治疗效应。为验证该假说,我们采用FDA批准的PARP1/2抑制剂奥拉帕利(Olaparib)及新一代具有血脑屏障穿透能力的PARP1选择性抑制剂AZD957441,在H3K27同基因的DMG细胞系及免疫完全的原位H3K27M突变DMG小鼠模型中开展研究。综合结果表明,PARP1/2抑制及选择性PARP1抑制不仅能增强H3K27M突变DMG的放射敏感性,还可协同放疗激活NK细胞介导的抗肿瘤免疫应答。
结 果
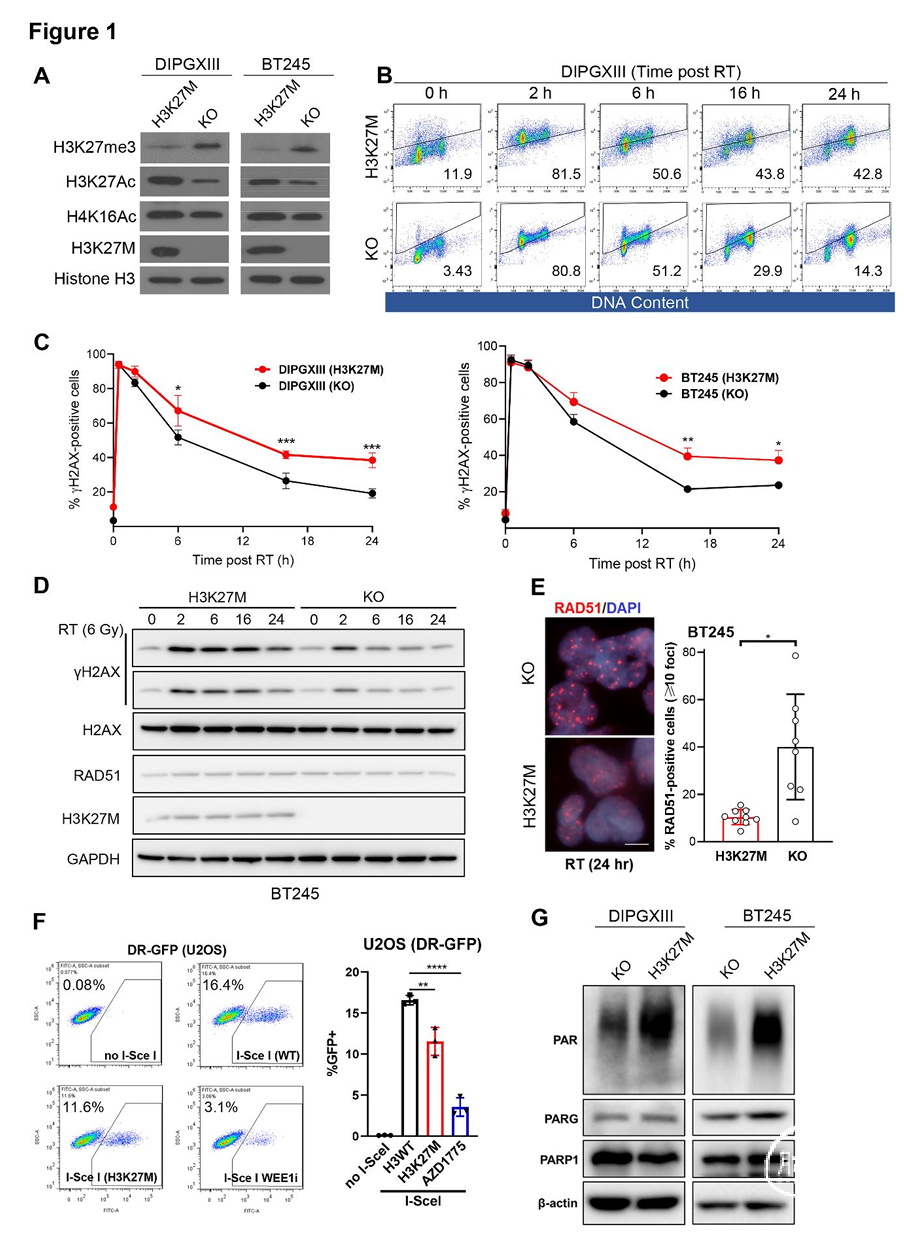
H3K27M突变细胞存在同源重组修复缺陷
H3K27M突变引起的基因表达异常可能导致DNA修复功能失调,但其对DNA修复的具体功能影响尚不明确。为揭示H3K27M突变导致的潜在DNA修复缺陷,我们利用CRISPR/Cas9基因编辑技术对携带自发H3K27M突变的同基因DMG细胞系进行处理,构建了保留一个H3.3野生型(WT)等位基因的敲除(KO)对照细胞(图1A)。
在原始状态及放疗处理后,H3K27M突变细胞相较于KO细胞均表现出更持久的γH2AX信号,提示H3K27M突变导致DNA修复缺陷(图1B-D;及补充图S1A、B,略)。此外,放疗诱导的G2/M期阻滞在H3K27M突变细胞与KO细胞中程度相当(补充图S1C,略)。DNA损伤信号的延迟动力学(16-24小时)提示同源重组修复(HRR)通路存在缺陷,因为HRR是相对缓慢的DNA双链断裂(DSB)修复途径。
为评估HRR活性,我们检测了HRR关键重组酶RAD51 foci的形成,发现H3K27M突变细胞在放疗后RAD51foci数量显著少于KO细胞(图1E)。与此一致,通过基于GFP的报告系统(DR-GFP)检测发现,携带H3K27M突变的U2OS细胞相较于野生型细胞HRR效率显著降低(图1F;及补充图S1D,略)。在H3K27M突变的BT245细胞中,HRR reporter系统活性也较同基因KO对照细胞显著下降(补充图S1E,略)。H3K27M突变DMG细胞对PARP抑制剂敏感性增强,进一步支持该突变导致功能性HRR缺陷(补充图S1F,略)。
与PARP抑制剂敏感性表型一致,H3K27M突变细胞的聚ADP核糖(PAR)水平高于KO细胞,而负责PAR合成与降解的关键酶PARP1和PARG的蛋白水平在两组间无差异(图1G)。
综上,H3K27M突变通过以下特征诱导HRR缺陷:放疗诱导DNA损伤持续存在、RAD51foci形成减少、HRR reporter活性降低,以及对PARP抑制剂敏感性增强。这些发现提示PARP可作为H3K27M突变型DMG的治疗靶点。
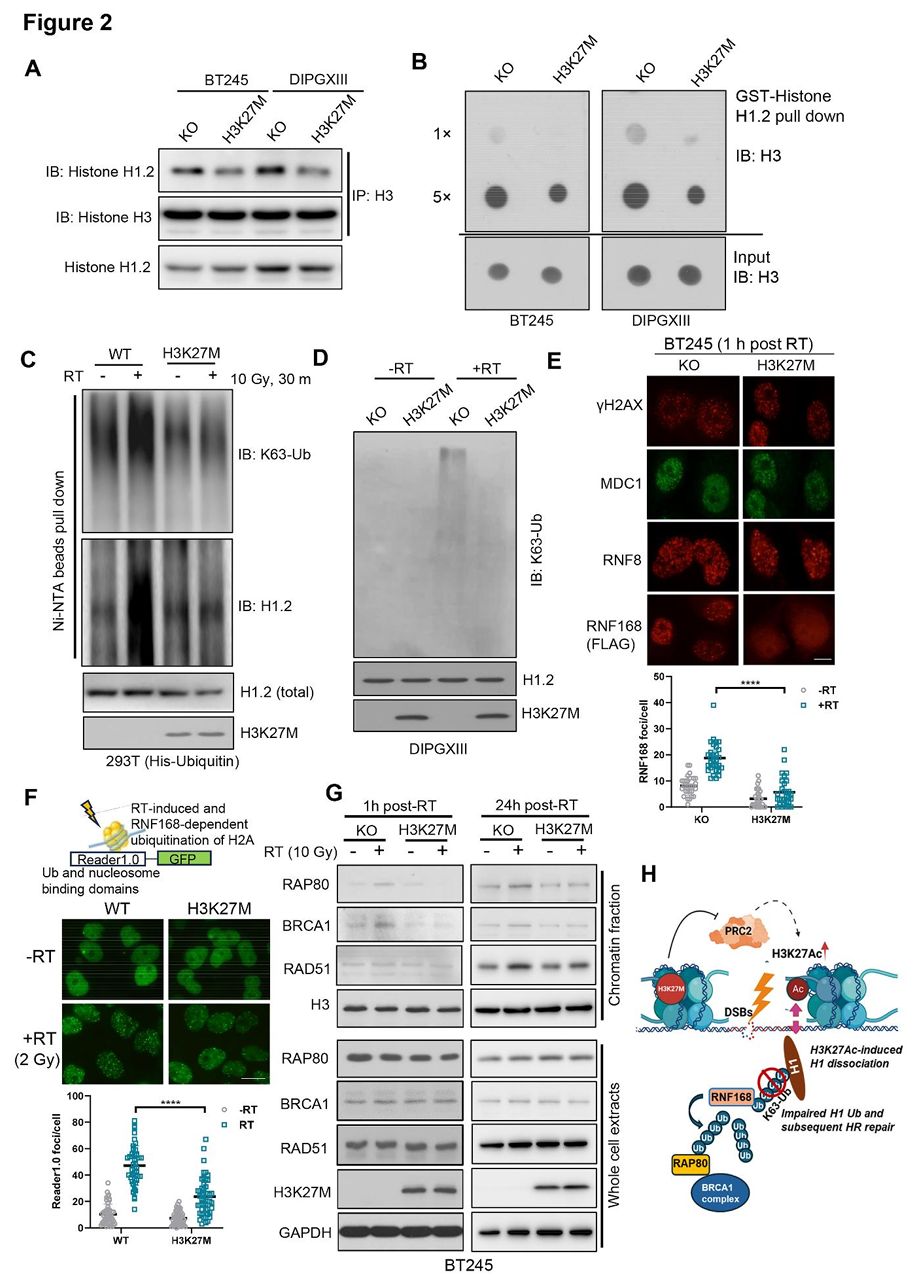
H3K27M突变通过阻断组蛋白H1的K63多聚泛素化抑制HRR蛋白募集
H3K27M突变导致显著的表观遗传失调和基因表达改变。为探究H3K27M细胞中HRR缺陷的机制,我们首先通过批量RNA测序分析了同基因H3K27M突变型BT245和DIPGXIII细胞中HRR相关基因的表达。出乎意料的是,两组同基因配对细胞系中HRR基因表达水平无显著差异(补充图S2A,略)。进一步分析儿童神经肿瘤联盟(PNOC)数据库发现,H3K27M突变型DMG患者与野生型儿童高级别胶质瘤(pHGG)患者的HRR基因表达水平相当,且H3K27M突变状态与HRR活性无相关性(补充图S2B、C,略)。这些结果表明,H3K27M介导的HRR缺陷并非由HRR基因表达改变引起。
组蛋白修饰通过促进DNA修复蛋白募集至损伤位点参与DNA损伤应答(DDR)。H3K27M作为功能获得性突变,导致全基因组H3K27me2/3标记缺失,同时核心组蛋白多个位点(如H3K27Ac和H4K16Ac)的乙酰化水平显著升高(图1A)。组蛋白高乙酰化可能阻碍连接组蛋白H1与染色质结合,而DNA损伤依赖的组蛋白H1非降解性泛素化(通过K63连接)对HRR蛋白(如BRCA1和RAD51)的募集至关重要。
实验显示,相较于同基因KO细胞,H3K27M DMG细胞中组蛋白H1.2与H3的结合减少(图2A)。利用纯化的GST-H1.2蛋白进行的体外结合实验进一步证实,H3K27M突变细胞中H1.2与H3的亲和力下降(图2B)。此外,H3K27M突变可阻断放疗诱导的H1.2 K63多聚泛素化在293T细胞中的积累(图2C)。与此一致,H3K27M突变型DMG细胞放疗后K63泛素化水平较KO细胞显著降低(图2D)。
DNA损伤时,E3泛素连接酶RNF8通过γH2AX/MDC1轴被募集至损伤位点,启动H1的K63多聚泛素化链形成。随后,RNF168通过其泛素相互作用模体(UIM)结合多聚泛素化的H1,进一步扩增损伤位点的K63泛素化信号。为明确H3K27M对γH2AX/MDC1/RNF8/RNF168轴的影响,我们检测了放疗后MDC1、RNF8和RNF168的焦点形成。结果显示,H3K27M与KO细胞中MDC1和RNF8焦点数量相当,但放疗后1小时H3K27M细胞中RNF168的募集显著减少(图2E),且该缺陷在放疗后24小时仍持续存在(补充图S2D,略)。
通过使用可特异性识别RNF168修饰的泛素化H2AK13/15位点的GFP融合报告系统(Reader1.0-eGFP),我们发现H3K27M突变细胞中Reader1.0向DNA损伤位点的募集显著减少(图2F),进一步支持RNF168在损伤位点的积累受损。RAP80(受体相关蛋白80)是一种泛素结合蛋白,可特异性识别K63多聚泛素链,从而引导BRCA1复合物定位至DNA损伤位点。实验表明,放疗后早期(1小时)和晚期(24小时),H3K27M突变型DMG细胞中RAP80/BRCA1/RAD51复合物向染色质的募集均存在缺陷(图2G)。
综上,H3K27M突变通过诱导核心组蛋白高乙酰化,导致H1与染色质解离,进而阻断DNA双链断裂位点HRR蛋白正确定位所需的K63泛素化信号,最终引发HRR缺陷(图2H)。
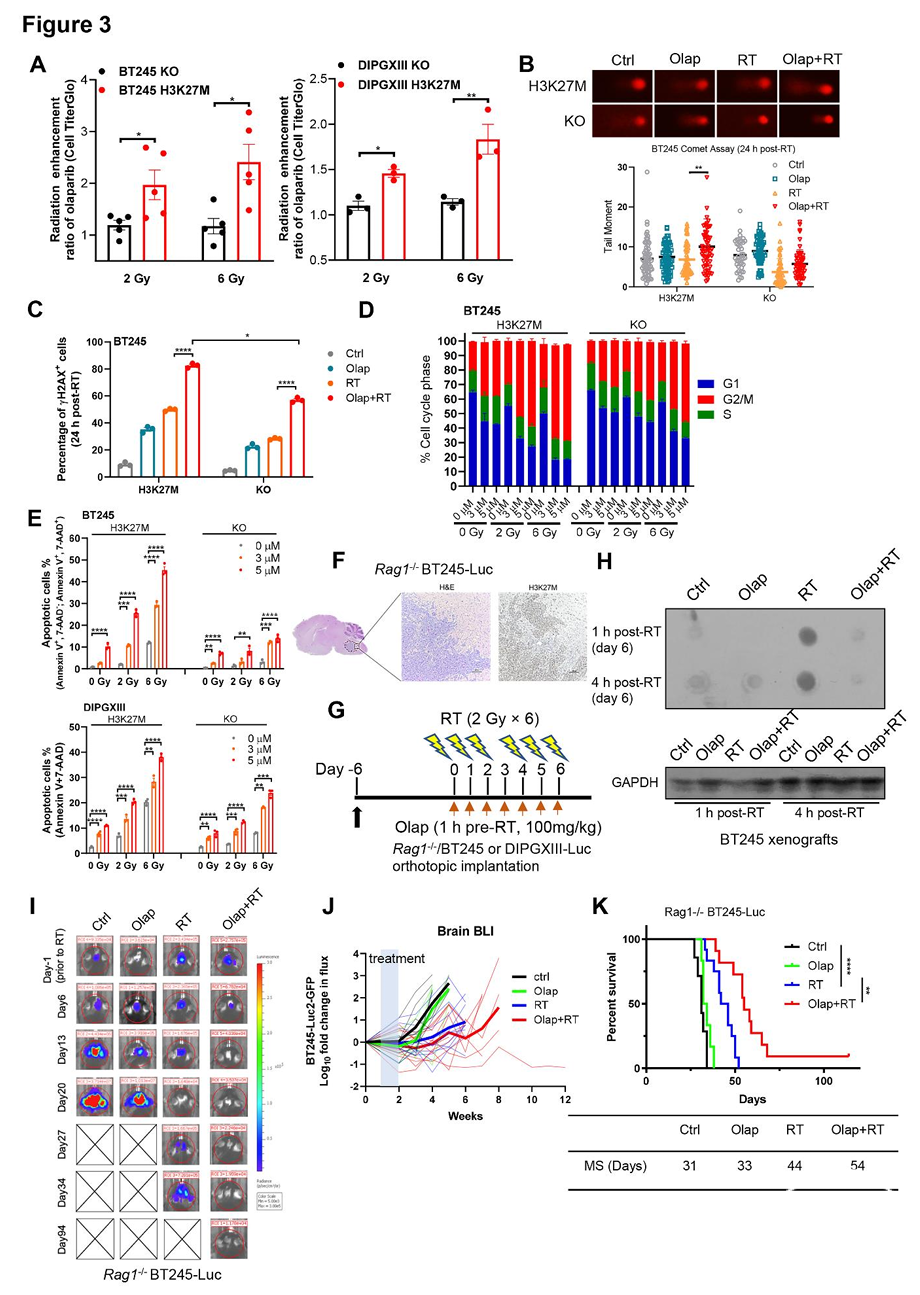
PARP抑制剂对H3K27M突变型DMG细胞、神经球及肿瘤具有选择性放疗增敏作用
PARP在修复放疗诱导的单链DNA断裂(SSBs,放疗损伤的主要类型)中至关重要。PARP抑制剂可使复制细胞中SSBs转化为细胞毒性双链断裂(DSBs),从而在HRR缺陷的癌细胞中引发合成致死效应。与此机制一致,奥拉帕利(olaparib)选择性增强人类(BT245和DIPGXIII)及小鼠(PPK,基因型p53R273H;PDGFRA;H3.3K27M)的放疗敏感性H3K27M突变型DMG细胞对低剂量(2 Gy)和高剂量(6 Gy)存在放疗敏感性,但对等基因KO或野生型(WT)细胞无此效应(图3A;及补充图S3A,略)。在神经球存活实验中,奥拉帕利同样增强了H3K27M突变型BT245和PPK细胞对放疗的敏感性(补充图S3B,略)。
相较于KO细胞,奥拉帕利联合放疗在H3K27M突变型DMG细胞中引起更持久的DNA损伤和G2/M期阻滞,表现为彗星尾矩增加、γH2AX信号持续以及G2/M期细胞比例升高(图3B-D;及补充图S3C-E,略)。此外,Annexin V/PI染色显示,奥拉帕利联合放疗在H3K27M突变细胞中诱导的凋亡程度显著强于KO细胞(图3E),而对细胞衰老无显著影响(补充图S3F,略)。
鉴于H3K27M突变对DMG肿瘤形成的必要性,我们选择H3K27M突变型DMG模型评估奥拉帕利联合放疗的体内疗效。通过螺丝导引法(方法示意图见补充图S3G,略),将携带荧光素酶标记的人源H3K27M突变型BT245或DIPGXIII细胞原位移植至Rag1-/-小鼠脑干。通过组织学检查和H3K27M免疫组化验证脑桥区移植瘤的形成(图3F)。
首先,我们评估奥拉帕利对原位脑肿瘤中PARP的抑制能力。奥拉帕利治疗显著降低了放疗后肿瘤组织中的PAR水平,表明药物可渗透至原位DMG肿瘤并有效抑制PARP活性(图3G、H)。为评价奥拉帕利联合放疗对H3K27M突变型DMG的疗效,我们通过生物发光成像监测BT245和DIPGXIII移植瘤的肿瘤负荷,并记录小鼠总生存期。在两种模型中,放疗或奥拉帕利联合治疗均显著降低肿瘤生物发光信号(图3I、J;及补充图S3H、I,略)。此外,联合治疗组小鼠的总生存期较单纯放疗组显著延长:BT245模型延长10天,DIPGXIII模型延长7天,且BT245模型中观察到10%的完全缓解率(图3K;及补充图S3J,略)。安全性分析显示,奥拉帕利联合脑干放疗耐受性良好,未引起显著体重下降或正常脑组织神经毒性(补充图S3K、L,略)。综上,本研究证实PARP抑制剂可选择性增强H3K27M突变型DMG对放疗的敏感性,且在体内展现出具有潜力的治疗指数。
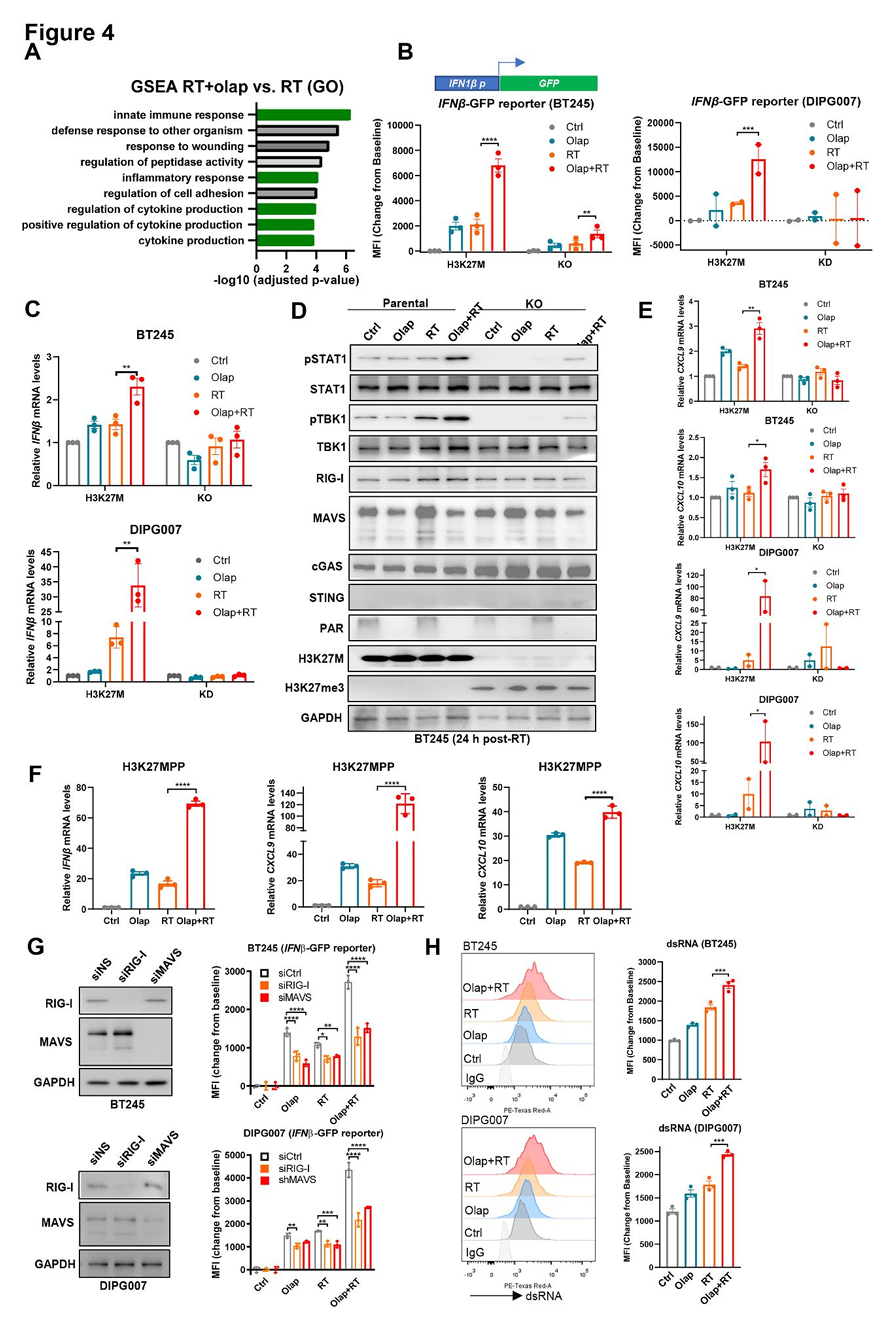
PARP抑制剂增强H3K27M突变型DMG细胞中放疗诱导的I型干扰素信号
放疗可增强肿瘤细胞的免疫原性及其对细胞毒性免疫细胞的可见性,而联合DDR抑制剂可进一步强化此效应。为全面解析PARP抑制剂联合放疗对H3K27M突变型DMG免疫原性的影响,我们进行批量RNA测序分析,发现显著富集的通路多与免疫相关,其中先天性免疫应答是奥拉帕利联合放疗(对比单纯放疗)最显著激活的通路(图4A;及补充表S1,略)。
鉴于I型干扰素(T1IFN)产生及信号传导在放疗诱导抗肿瘤免疫中的关键作用,我们在H3K27M突变型DMG细胞及其同基因敲除(KO)或敲低(KD)对照细胞中构建了IFNβ1启动子驱动的GFP reporter系统(图4B)。单独使用奥拉帕利或放疗仅轻微诱导IFNβ1-GFP报告基因表达,而联合治疗在H3K27M突变细胞中表现出协同激活效应,且显著强于对照细胞(图4B)。与报告基因结果一致,奥拉帕利联合放疗选择性上调H3K27M突变细胞中内源性IFNβ1和IFNα4 mRNA表达,其中DIPG007细胞中IFNβ1的诱导倍数超过100倍(图4C;及补充图S4A,略)。
T1IFN信号激活的分子标志物分析显示,奥拉帕利联合放疗显著增加T1IFN信号通路上游磷酸化TBK1(S172)及下游磷酸化STAT1(Y701)水平,且此效应在H3K27M突变细胞中最为显著(图4D;及补充图S4B,略)。此外,干扰素刺激基因CXCL9和CXCL10的表达在联合治疗后亦显著上调(图4E)。为验证上述发现,我们在小鼠H3K27M DMG模型(H3K27MPP,由表达H3K27M、p53R273H及PDGFR的神经祖细胞构建)中开展实验,结果显示与人源模型一致:奥拉帕利联合放疗显著诱导IFNβ1、CXCL9和CXCL10 mRNA表达,并伴随p-TBK1和p-STAT1的轻度激活(图4F;及补充图S4C,略)。
进一步探究T1IFN诱导的分子机制发现,DMG细胞中cGAS/STING通路关键蛋白(cGAS、STING)表达缺失,而RIG-I/MAVS通路蛋白表达显著(图4D;及补充图S4B,略)。敲低RIG-I或MAVS可显著抑制放疗联合奥拉帕利诱导的T1IFN报告基因活性,表明RIG-I/MAVS介导的双链RNA(dsRNA)识别通路在此过程中起核心作用(图4G)。通过dsRNA特异性J2抗体免疫荧光染色,我们观察到细胞质内dsRNA聚集,且经dsRNA特异性RNA酶III处理后信号消失,证实J2抗体的特异性(补充图S4D,略)。流式细胞术定量分析显示,放疗可增加BT245和DIPG007细胞中dsRNA水平,而奥拉帕利联合治疗进一步强化此效应(图4H)。
PARP抑制剂联合放疗诱导NKG2D活化配体表达并增强NK细胞介导的肿瘤裂解
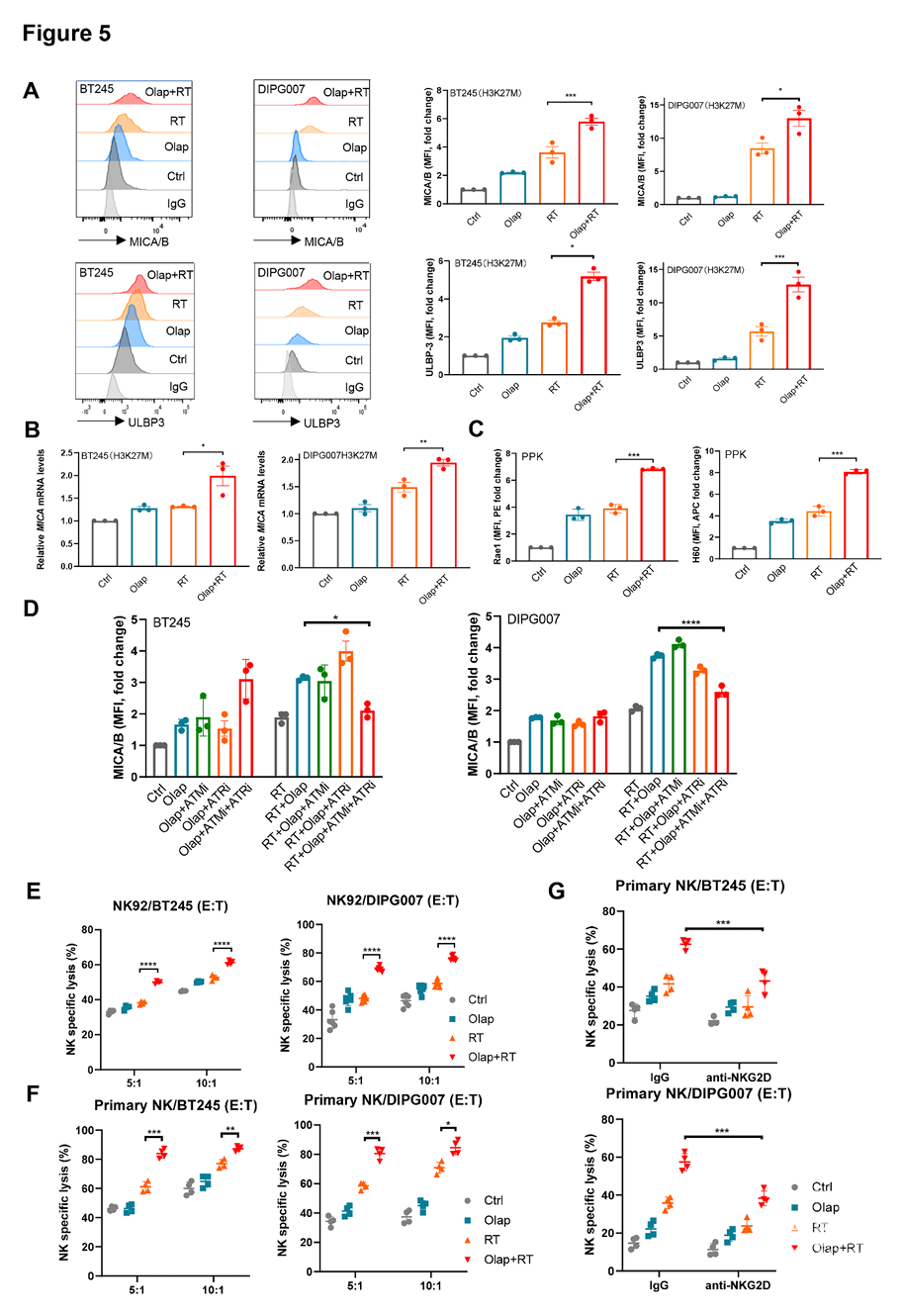
I型干扰素(T1IFN)对T细胞和NK细胞的扩增、成熟及活化至关重要。尽管细胞毒性T细胞在抗肿瘤免疫中起核心作用,但NK细胞在DMG中展现出更强的杀伤潜力,且其活性与患者总生存期相关。NK细胞的关键功能特征是通过NKG2D等活化受体识别应激态肿瘤细胞表面配体(人源为MICA、MICB、ULBP1-6;鼠源为Rae1、H60、Mult1),进而触发脱颗粒、细胞因子分泌及靶细胞裂解。
为评估NK细胞在奥拉帕利联合放疗疗效中的作用,我们首先检测DMG肿瘤中表达的NKG2D活化配体MICA/B和ULBP-3。联合治疗使H3K27M突变型DMG细胞表面MICA/B和ULBP-3表达上调5-10倍(图5A),并显著增加其mRNA水平(图5B;及补充图S5A,略)。在小鼠H3K27M突变型PPK DMG模型中,类似配体Rae1和H60的表达亦被联合治疗上调(图5C)。此效应具有PARP抑制特异性,因联合ATM或ATR抑制剂与放疗时NKG2D配体表达未被增强(补充图S5B,略)。值得注意的是,与既往研究一致,奥拉帕利联合放疗诱导的NKG2D配体表达依赖ATM和ATR活性,同时抑制二者可阻断BT245和DIPG007细胞中MICA/B的诱导(图5D)。
此外,联合治疗对NKG2D配体的诱导不依赖T1IFN信号,因T1IFN受体阻断抗体未能减弱其表达(补充图S5C,略)。NK细胞活性亦受抑制性配体调控,我们检测了与NKG2A/p94及杀伤细胞免疫球蛋白样受体(KIRs)结合的抑制性配体HLA-E和HLA-A/B/C,发现联合治疗未诱导其表达(补充图S5D,略)。PARP抑制剂联合放疗通过上调H3K27M突变型DMG细胞表面NKG2D活化配体,同时不改变抑制性配体水平,促使NK细胞调控信号向激活方向偏移,从而增强其抗肿瘤活性。
为评估奥拉帕利联合放疗是否增强NK细胞对H3K27M突变型DMG细胞的杀伤,我们将经联合处理的BT245和DIPG007细胞用钙黄绿素AM染料标记,随后与人NK-92效应细胞(FDA批准的淋巴瘤来源NK免疫治疗细胞系)以5:1和10:1的效靶比(E:T)共培养。联合治疗显著增加两种DMG细胞系的裂解率(图5E)。进一步使用原代NK细胞(补充图S5E)验证发现,无论高低效靶比下,经PARP抑制剂和放疗处理的DMG细胞均表现出更强的NK介导杀伤(图5F;及补充图S5F,略)。
NK细胞对DMG细胞的杀伤可被NKG2D阻断抗体逆转,证实该过程依赖NKG2D活化(图5G)。为探究NK细胞杀伤的肿瘤选择性,我们在正常人星形胶质细胞(NHA)中开展平行实验。结果显示,放疗后PARP抑制剂未上调NHA细胞的MICA/B表达,亦未增强NK细胞对其杀伤(补充图S5G-H,略)。综上,PARP抑制剂联合放疗通过特异性上调H3K27M突变型DMG细胞中NK活化配体,促使活化NK细胞选择性裂解肿瘤细胞,而对正常细胞无显著影响。
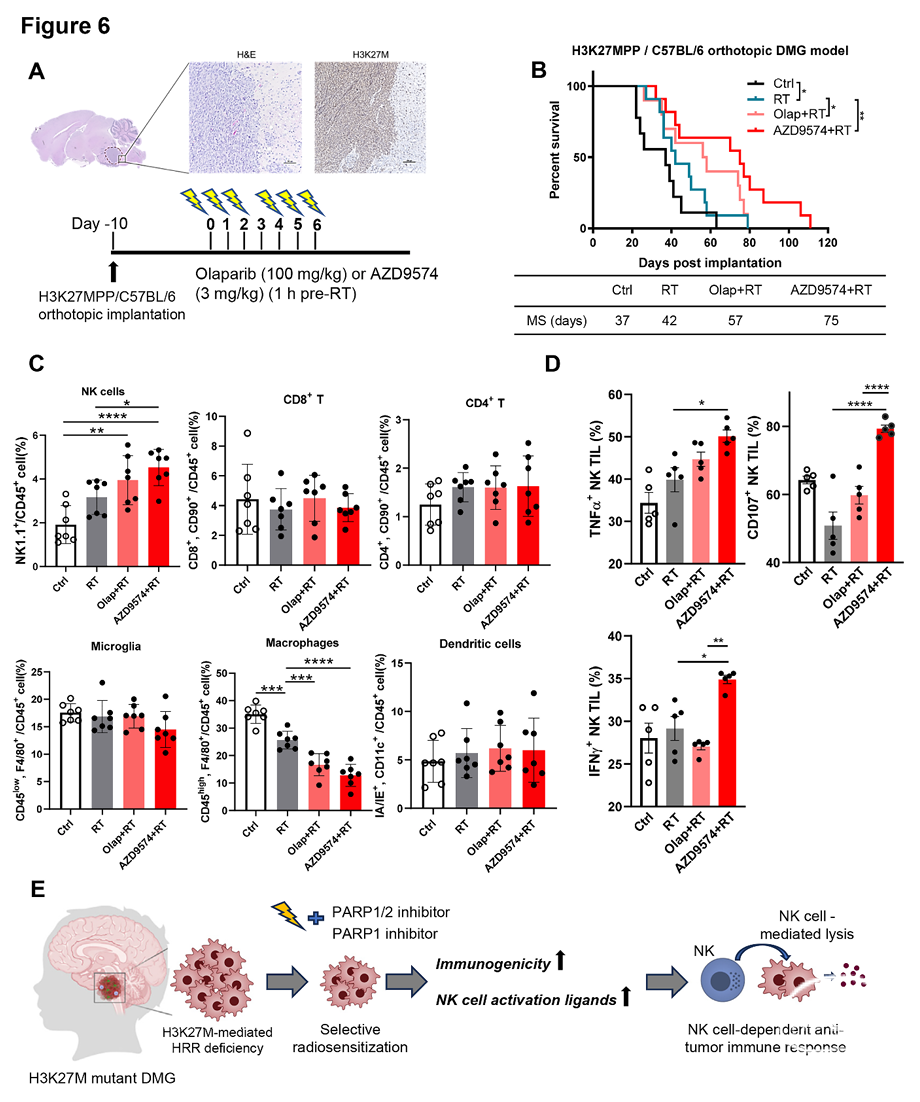
PARP1选择性抑制剂联合放疗增强H3K27M突变型DMG小鼠模型瘤内NK细胞活性并延长生存期
为探究免疫系统(尤其是NK细胞)在H3K27M DMG治疗反应中的作用,我们构建了免疫完全的同源原位DMG模型:将H3K27MPP细胞植入C57BL/6小鼠脑干(图6A)。为优化治疗策略,我们引入新一代PARP1选择性抑制剂AZD9574(具有血脑屏障穿透性)。荷瘤小鼠分别接受单放疗(RT)、RT联合奥拉帕利或AZD9574治疗(图6A)。结果显示,单独放疗组组中位生存期从37天延长至42天,而奥拉帕利联合RT进一步显著延长至57天。值得注意的是,AZD9574联合RT疗效最佳,中位生存期达75天,且未出现体重下降或脑毒性(图6B;及补充图S6A-B,略)。随后,我们分析瘤内免疫细胞亚群(补充图S6C,略),发现基线水平下原位DMG肿瘤中已存在NK细胞(补充图S6D,略)。治疗后第1天,RT增加瘤内NK细胞比例(CD4+和CD8+ T细胞无变化),联合PARP抑制剂后该效应进一步增强(图6C)。此外,RT联合PARP抑制剂显著降低瘤内巨噬细胞(CD45 high/CD11b+/F4/80+)比例(图6C)。进一步功能检测表明,PARP抑制剂联合RT显著提升瘤内NK细胞活性(TNFα+、CD107+或IFNγ+标志物),但对CD8+ T细胞活化状态无影响(图6D;及补充图S6E-F,略)。NK细胞耗竭实验证实其在治疗反应中的关键作用(补充图S6G,略)。值得注意的是,AZD9574联合RT激活瘤内NK细胞的效果优于奥拉帕利,可能归因于其更强的脑穿透性和PARP1选择性(图6D)。综上,本研究提出机制模型(图6F):PARP抑制剂联合放疗通过激活H3K27M DMG肿瘤微环境(TME)中NK细胞的细胞毒性功能发挥治疗作用。选择性PARP1抑制剂AZD9574因优化特性,疗效显著优于PARP1/2双抑制剂奥拉帕利。
讨 论
H3K27M突变组蛋白是DMG的致癌驱动因子,但并非直接治疗靶点。该突变通过破坏核心组蛋白正常功能,导致DNA修复异常。本研究发现,H3K27M突变引起核心组蛋白过度乙酰化,促使连接组蛋白H1解离,进而抑制H1的K63多聚泛素化及RNF168/BRCA1/RAD51蛋白募集,最终导致同源重组修复(HRR)缺陷。这一发现揭示了H3K27M突变型DMG中核心组蛋白与连接组蛋白H1的功能互作,并印证了染色质结构与组蛋白修饰在HRR中的关键作用。
除影响RAP80/BRCA1招募外,53BP1的重定位依赖于RNF168介导的组蛋白H2A/H2AX在K13/15位点的泛素化。H3K27M突变细胞中RNF168灶点形成失败及H2A/H2AX(K13/K15)泛素化缺陷,与既往研究发现的53BP1募集受损现象一致,共同支持H3K27M突变对DNA修复途径的深远影响。
H3K27M突变引起的HRR缺陷可部分解释DMG患者对放疗的初始敏感性,但其复发率高于H3K27野生型高级别胶质瘤,且生存结局更差。本研究证实,H3K27M介导的HRR缺陷可作为治疗突破口,通过PARP抑制剂增强放疗敏感性。H3K27M突变亦增加DMG细胞对PARP抑制剂单药的敏感性(图S1F,略),但体内实验中该效应未重现,可能因本研究的放疗增敏方案给药周期较短,而HRR缺陷肿瘤中PARP抑制剂单药需长期暴露方可显效。
除HRR缺陷相关的协同作用外,DMG患者肿瘤组织中PARP1表达显著高于正常组织,且H3K27M突变细胞中多聚ADP核糖(PAR)水平高于野生型,进一步支持PARP靶向治疗策略的可行性。然而,临床研究中PARP1/2抑制剂Veliparib联合放疗未改善DMG患者生存。值得注意的是,该试验中大部分患者外周血单核细胞(PBMCs)的PAR活性未因Veliparib治疗下降,且PAR降低与生存无相关性。尽管失败原因尚未明确,但药物脑穿透性不足及PARP捕获效能较弱(下文讨论)可能是关键因素。
本研究显示,在激活NK细胞及延长同源原位模型生存期方面,新一代血脑屏障穿透性PARP1选择性抑制剂AZD957441优于第一代PARP1/2抑制剂奥拉帕尼。PARP1抑制与PARP1/2抑制的疗效差异可能源于以下几个因素:首先,PARP1是细胞聚ADP核糖基化(PARylation)的主要介导者,负责绝大多数DNA损伤诱导的PARylation过程;其次,PARP抑制剂(如奥拉帕尼和AZD9574)引发的PARP捕获效应(即PARP1滞留于DNA损伤位点)是放射增敏作用(以及单药疗效)的关键机制。相比之下,PARP2可能并非抗癌活性所必需,且在造血细胞存活及维持细胞毒性T细胞和NK细胞活性中起关键作用。除对NK细胞的调控作用外,PARP抑制剂还导致放疗后肿瘤微环境(TME)中巨噬细胞数量减少。鉴于包括巨噬细胞和小胶质细胞在内的疾病相关髓系细胞(DAM)构成DMG中主要的非肿瘤细胞群体并发挥促瘤作用,观察到的巨噬细胞减少现象支持PARP抑制剂联合放疗的协同治疗效应。未来研究将通过单细胞RNA测序、空间分子成像分析等技术手段,系统解析上述机制对PARP抑制剂联合放疗疗效的相对贡献,重点关注H3K27M突变型DMG肿瘤微环境中的免疫调节效应。
我们与其他研究团队已证实,通过激活内源性核酸感应通路,抑制DNA损伤修复(DDR)可进一步增强放疗诱导的免疫原性DNA损伤,从而提升肿瘤免疫原性。本研究发现,相较于同源基因敲除(KO)对照组,亲本H3K27M突变型DMG细胞在PARP抑制剂联合放疗后更显著地诱导I型干扰素(T1IFN)表达及信号传导,这可能归因于突变细胞中开放的染色质结构及免疫原性DNA损伤。I型干扰素主要通过刺激CD8+ T细胞和NK细胞等免疫细胞来发挥抗肿瘤作用。尽管DMG肿瘤细胞表达主要组织相容性复合体I类(MHC-I)分子,但相较于细胞毒性T细胞,NK细胞在DMG中展现出更强的细胞毒活性潜力。放疗及其他基因毒性药物能有效诱导肿瘤细胞表面NK细胞活化配体(调控NK细胞上的NKG2D受体)的表达。值得注意的是,这种诱导作用依赖于ATM/ATR/CHK1/2信号通路,这使PARP抑制剂在增强放疗诱导的NKG2D配体表达及NK细胞杀伤效应方面具有独特优势。研究发现,抗体介导的NKG2D阻断可消除PARP抑制剂联合放疗后NK细胞对DMG细胞的杀伤作用,表明NKG2D配体诱导对NK细胞杀伤效能至关重要。尽管PARP抑制剂联合放疗诱导NKG2D配体表达的过程不依赖T1IFN信号通路,但这些信号轴仍可能协同介导抗肿瘤免疫反应。
同源原位移植的H3K27M突变型DMG肿瘤微环境(TME)无法完全模拟人类DMG的特征——后者以淋巴细胞匮乏的免疫沉默型微环境为特点。使用基因工程构建的H3K27M突变型DMG模型可能是更具临床相关性的研究体系,有助于理解PARP1及PARP1/2抑制剂联合放疗对TME的影响。另一方面,NK细胞导向的免疫治疗(如CAR-NK细胞疗法)为克服DMG的免疫静默型TME提供了替代策略。通过PARP抑制剂联合放疗对H3K27M突变DMG肿瘤细胞进行选择性预激活,可能进一步强化此类疗法的效果。
本研究揭示了H3K27M突变与同源重组修复(HRR)缺陷之间的新关联,这种缺陷导致PARP抑制可选择性增强H3K27M突变型DMG的放射敏感性(图6E)。放射增敏效应与先天免疫反应增强以及DMG细胞中NK细胞活化配体表达增加相关。与免疫原性提升的表现一致,PARP抑制剂联合放疗能促进NK细胞对DMG肿瘤细胞(而非正常星形胶质细胞)的特异性杀伤,在免疫完全的DMG肿瘤模型中产生治疗反应,并增加NK细胞的数量和活化状态——这些改变是治疗起效的必要条件。最后,本研究支持PARP1选择性抑制剂相较于PARP1/2抑制剂可能提供更优的治疗指数,为PARP1抑制剂联合放疗在H3K27M突变型DMG中的未来临床开发铺平道路,同时也为后续与NK细胞疗法或免疫治疗的联合应用提供了可能性。
第一作者简介

郭育培
● 博士研究生,主要研究方向为脑胶质瘤的基础与临床治疗
● 参与多项国家自然科学基金面上项目,主要参与“儿童H3K27M突变型脑桥胶质瘤中PARP抑制剂联合放疗增强B7-H3靶向CAR-NK免疫治疗的作用研究机制”
● 以第一作者发表SCI论文3篇(JCR分区:一区1篇;二区1篇;三区一篇)。单篇最高影响因子16.4分

李姿安
● 三年级博士研究生,主要研究方向为GBM与DMG的基础与临床研究
● 参与国家自然科学基金面上项目“脑胶质瘤中泛素连接酶Cullin5-SOCS3通过泛素化降解膜蛋白CMTM6来调控PD-L1蛋白水平”、“儿童H3K27M突变型脑桥胶质瘤中PARP抑制剂联合放疗增强B7-H3靶向CAR-NK免疫治疗的作用研究机制”
● 截止目前共参与发表SCI论文12篇,其中第一作者及共同一作SCI论文3篇,总影响因子38.6分
通讯作者简介

王新军 教授
郑州大学第三附属医院
● 医学博士,二级教授、主任医师,博士生导师,中原名医
● 河南省医学重点学科神经外科学科带头人;河南省胶质瘤代谢与微环境研究国际联合实验室主任;河南省儿童脑胶质瘤精准诊疗工程研究中心主任。代谢紊乱与食管癌防治全国重点实验室临床PI。郑州大学发育与代谢研究院主任
● 河南省医师协会神经外科医师分会会长,中华医学会神经外科分会全国委员、中国医师协会神经外科分会全国常委
● 长期从事脑肿瘤及颅脑损伤的诊断治疗、临床教学及科研工作。主持参与国家自然科学基金4项,承担国家重点研发计划(子项)3项、省部级基金10项。第一或通讯作者发表中英文论文100余篇。主编参编专著与教材5部
● 王新军团队目前有高级职称9名,中级职称25名;博士生导师2人,硕士生导师8人,在读硕士21人,在读博士10人;博士后10人
主要研究方向,代谢重塑与神经肿瘤防治研究:
● 在国际上首次阐述了FBXW7在胶质瘤患者中可作治疗的潜在靶点,获得2022年度中国神经外科领域高价值论文TOP100;建立了具有河南省特色的儿童中枢神经系统肿瘤生物样本信息数据库和河南省妊娠相关性脑卒中患者和子代队列;
● 在儿童脑胶质瘤临床和研究方面,开展我国首个针对儿童弥漫性内生型脑桥胶质瘤(DIPG)的PARP抑制剂临床试验;拥有原代细胞、类器官、CDX、PDX、自发诱导模型等基础与临床应用平台,已经开展针对弥漫性中线胶质瘤患者的治疗窗口期快速药效学评价研究;已获得国家自然基金面上项目“儿童H3K27M突变型脑桥胶质瘤中PARP抑制剂联合放疗增强B7-H3靶向CAR-NK免疫治疗的作用机制研究”、河南省国际科技合作重点项目“PARP抑制剂对H3K27M突变型儿童弥漫性内生型脑桥胶质瘤(DIPG)放疗增敏的安全性、有效性研究”以及河南省科技攻关项目“尼拉帕利靶向药物设计及联合放疗治疗儿童弥漫性内生型脑桥胶质瘤的研究”;河南省医学科技人才海外研修项目“通过PARP抑制剂和放疗联合治疗改善NK细胞介导的针对H3K27M突变型DIPG的抗肿瘤免疫反应”;国家自然科学基金青年项目“肿瘤细胞外囊泡介导小胶质/巨噬细胞代谢重编程抑制弥漫性中线胶质瘤对ONC201敏感性的机制研究”、“NBS1通过SUMO修饰调控同源重组修复的机制研究”和“RNF7调控DNA损伤应答与胶质母细胞瘤放化疗敏感性的机制研究”;国家博士后科学基金面上项目“FNTA通过异戊二烯化RAP1B促进胶质瘤增殖的作用机制研究”和“RNF7调控DNA损伤应答与胶质母细胞瘤放化疗敏感性的机制研究”等。
声明:脑医汇旗下神外资讯、神介资讯、神内资讯、脑医咨询、Ai Brain 所发表内容之知识产权为脑医汇及主办方、原作者等相关权利人所有。
