




提示
前言
神经肿瘤是颅内常见病,主要包括神经上皮肿瘤、脑膜瘤、转移瘤及淋巴瘤等。从483期开始将刊发“第二轮神经肿瘤系列”,与同道共享,欢迎大家批评指正和交流讨论。
病例简介
患者,女,59岁,因“头痛伴恶心呕吐,记忆力减退5天”经当地医院CT检查,诊断为“三脑室肿瘤”来我院门诊后收入院。
入院查体:神清,精神软,反应迟钝,对答部分切题,部分遵嘱动作,远近记忆力减退,时间定向力,空间定向力减退,颅神经检查无殊。四肢肌力及肌张力正常,神经系统生理反射存在,病理反射未引出。
诊疗经过
患者入院后完善相关检查,头颅CT提示:三脑室等密度占位,伴侧脑室扩张,脑积水(图1)。进一步完善头颅增强MRI示:鞍上及三脑室内占位,继发脑积水伴间质性水肿,考虑脊索样胶质瘤可能(图2),同时行垂体激素检测,部分激素降低(图3)。结合患者病情及检查结果,考虑为“三脑室肿瘤(性质?),脑积水”。
科室讨论认为手术指征明确,逐于2025-03-25在全麻下经纵裂胼胝体入路行第三脑室肿瘤切除术,术中见肿瘤位于胼胝体穹窿下,呈灰白色,鱼肉胶冻样,质地稍韧,肿瘤与周围脑组织粘连(图4),术中冰冻提示:黏液背景中见肿瘤细胞条索样排布,考虑脊索样胶质瘤(图5)。显微镜下瘤内减压后,小心分离肿瘤边界,分块逐步切除肿瘤。
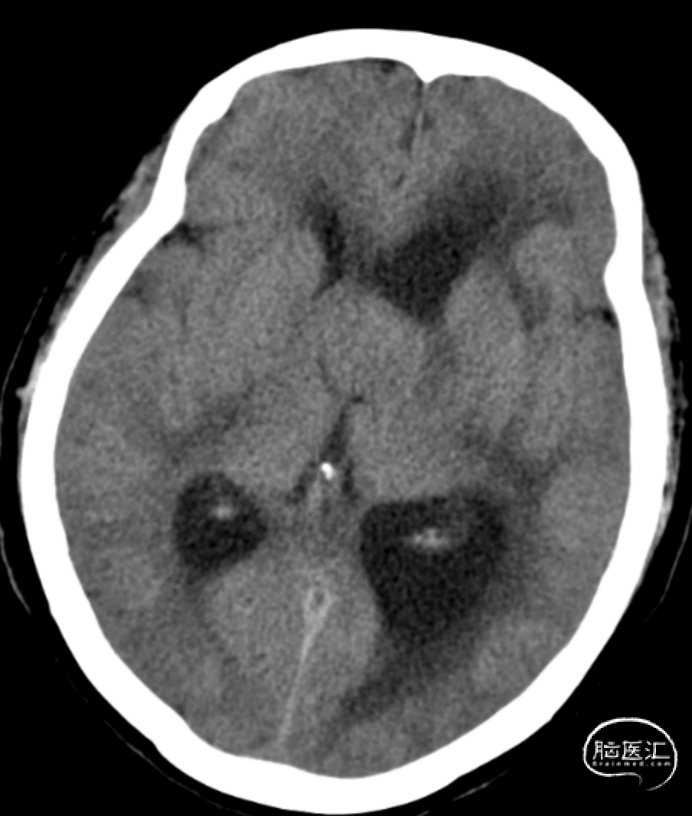
图1. 头颅CT平扫示:三脑室等密度占位,大小约28.8*21.7.*30.5mm,内未见钙化,伴侧脑室扩张,脑积水表现。

图2. 头颅增强MRI示:鞍上及三脑室内占位,混杂长T1长T2信号,大小约28.6*22.5*30.9mm,界尚清,增强明显不均匀强化,DWI呈稍低信号,灶周见水肿影。两侧额叶皮层下、侧脑室旁、基底节区、半卵圆中心见多发点片状长T2高信号,脑室系统扩张,中线稍右移。
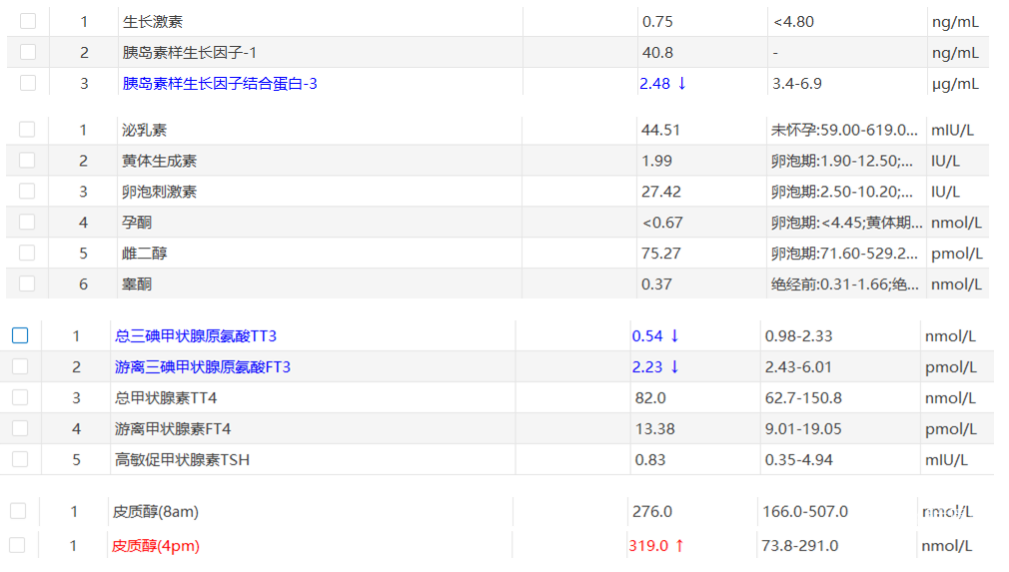
图3. 垂体激素检测结果。

图4.术中可见肿瘤位于穹窿下,灰白色,鱼肉胶冻样,质地稍韧。
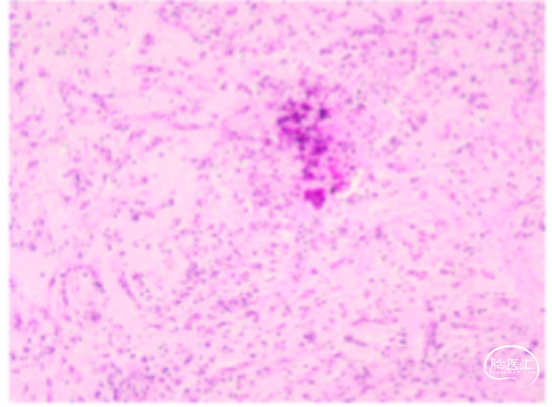
图5.术中冰冻示:黏液背景中见肿瘤细胞条索样排布,考虑脊索样胶质瘤。
术后继续脑室外引流,同时予以脱水降压,预防癫痫,抗感染,护胃止吐,适当补液等对症处理,并密切监测电解质和激素水平。术后第1天复查头颅CT(图 6)见术区少许出血,术后第12天(图 7)复查头颅增强MRI提示肿瘤全切,颅内情况稳定。患者术后一般情况可,神清,反应可,对答尚可,GCS15分,四肢查体配合。术后恢复良好,出院。
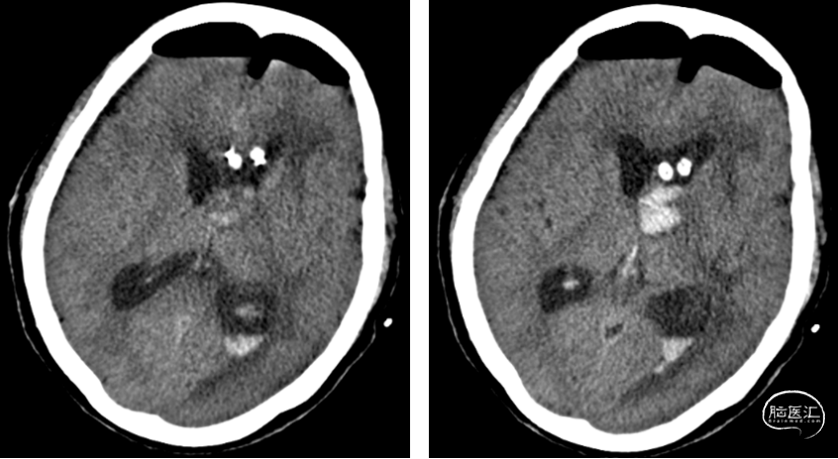
图6. 开颅肿瘤切除术后第一天复查头颅CT呈术后改变,术区少量积血积液,颅内积气。
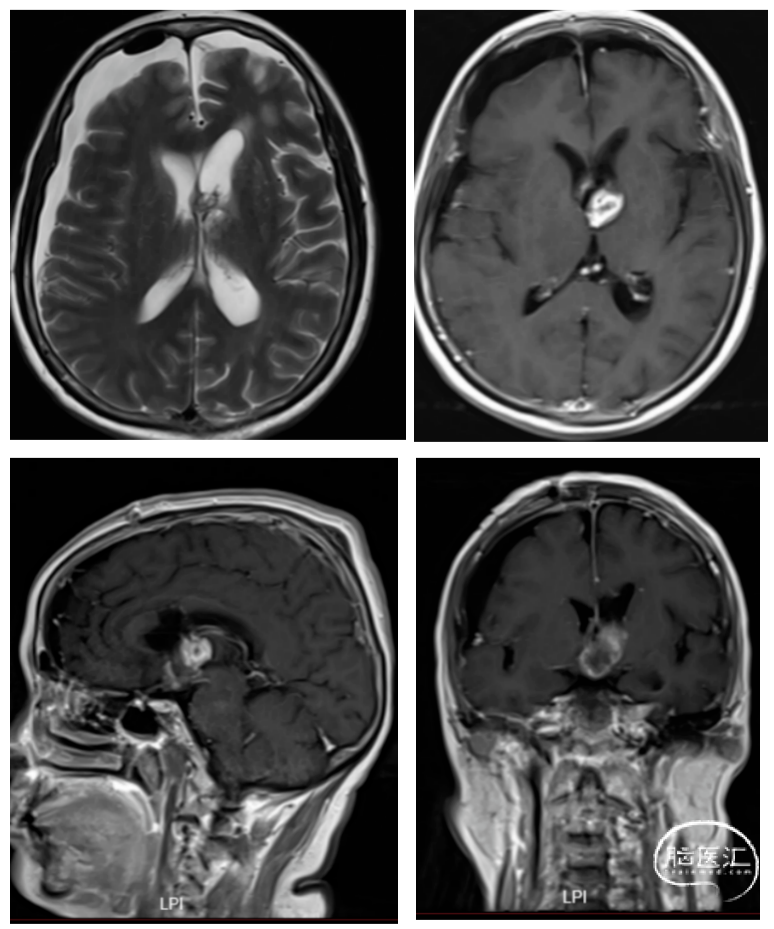
图7.术后MRI显示肿瘤全切。
病理结果
常规病理报告:(鞍上)粘液背景中见肿瘤细胞条索样排列,伴多量淋巴细胞浸润,考虑脊索瘤样胶质瘤, CNS WHO 2级,建议NGS检测进一步明确(图8)。
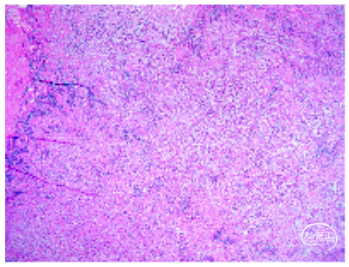
图8. (鞍上)粘液背景中见肿瘤细胞条索样排列,伴多量淋巴细胞浸润,考虑脊索瘤样胶质瘤。
免疫组化:A6-06:GFAP(+), Olig2(-), EMA(+), SSTR2(+,弱), PR(-), STAT6(-), CK-PAN(-), S100(+,散在), Brachyury(-), α-SMA(-), Desmin(-), ALK(+,浆), CD31(+), CD34(+), Ki-67(1-2%,+), ERG(-), INI1/SMARCB1(+), SMARCA4/BRG1(+), TTF-1(SPT24)(+)。
讨论
脊索样胶质瘤(Chordoid Glioma)是一种罕见的中枢神经系统肿瘤,同时具有神经胶质细胞和脊索样特征。这种肿瘤最初被Wanschitz等人于1995年描述为一种独特的脑膜瘤变异型[1],1998年Brat首次将其正式命名为脊索样胶质瘤[2]。在2021年的第五版中枢神经系统肿瘤分类中,脊索样胶质瘤被归属于局限性星星胶质细胞瘤。目前关于脊索样胶质瘤的组织起源仍存在争议:Sato等人认为神经胶质细胞为其起源,即脊索样胶质瘤起源于第三脑室前部的室管膜细胞[3];Suh等报道了一例鞍上脊索样胶质瘤合并Rathke囊肿的病例,他们认为脊索样胶质瘤可能起源于Rathke囊的多能干细胞[4];基于脊索样胶质瘤具有室管膜的超微结构特征,且其多位于第三脑室,Cenacchi等人认为脊索样胶质瘤可能是室管膜瘤的特定亚型[5]。Bielle等人在17例第三脑室脊索样胶质瘤中发现了恒定的TTF-1免疫标记,因而认为其可能起源于终板的血管[6]。因此,将来需要进一步证据来明确脊索样胶质瘤的确切来源。脊索样胶质瘤先前一直被认为仅发生在第三脑室及其延伸的鞍上区和侧脑室。近年来有文献报道了位于第三脑室外的脊索样胶质瘤:Goyal等人于2007年报道了一例顶颞叶脑室外脊索样胶质瘤[6],Jain等人于2008年报道了一例累及丘脑右放射冠状脑室旁的脊索样胶质瘤[7],Kim等人于2010年报道了一名丘脑脊索样胶质瘤患者。脊索样胶质瘤多发生于成年人,女性多于男性女性[8]。一项对98例患者的统计数据显示,脊索样胶质瘤男女比例约为1:1.7(36名男性和62名女性),患病年龄为30-60岁 (n=80/98,81.6%),仅4例儿童患者[9]。
脊索样胶质瘤起病缓慢,病程较长,除可出现头痛、头晕、失眠、乏力、嗜睡等神经症状和精神障碍外,其临床表现还与肿瘤发生的位置、大小和压迫的组织密切相关。如前所述,脊索样好发于第三脑室前部,并向鞍上和侧脑室延伸,因而最常见的临床表现为肿瘤阻塞第三脑室或中脑导水管引起的梗阻性脑积水,当下丘脑受累时可引起下丘脑功能障碍(包括内分泌激素紊乱和/或电解质失调、尿崩症、体温调节异常等)[10],当肿瘤体积较大引起视神经压迫时可表现出相应的视力下降或视野缺失[11]。由于脊索样胶质瘤临床表现不具有特异性,因而易被误诊为其他第三脑区肿瘤(如颅咽管瘤、垂体瘤)。本例病例中,患者并无特征性症状,仅表现为颅内高压,出现恶心呕吐,并伴发记忆里减退。
据文献报道脊索样胶质瘤在影像学上常具有一定的特征:1.肿瘤多位于第三脑室前部,呈类圆形,边界尚清晰,可向周围延伸,累及鞍上池和下丘脑,合并幕上脑室梗阻性脑积水[6,12]。2.肿瘤内部无明显囊变、出血和坏死,也少见钙化,因而CT和MR上多信号均匀,CT上可表现为等密度或稍高密度影,MR上T1WI多呈低信号,T2WI可呈等或高信号,常表现为高信号,这可能与肿瘤细胞和粘液基质的比例相关,增强后肿瘤常显著强化,多表现明显均一强化[13]。当肿瘤较大累及视束时,可出现对称性分布的血管源性水肿[9]。本例患者影像学特征典型,肿瘤位于鞍上及三脑室,边界尚清,并继发脑积水伴间质性水肿,CT呈等信号,MR上T1低信号,T2稍高高信号,增强明显,不均匀强化。这很好的指导了我们对于手术方案的选择。
脊索样胶质瘤组织学特征为肿瘤细胞呈上皮样,排列成条索状或巢状。成簇或散在分布在黏液样基质中,肿瘤细胞核圆形或类圆形,核小深染,无明显核分裂像,富含嗜酸性胞质。肿瘤组织和周围组织分界常清楚,间质可有淋巴细胞和浆细胞浸润以及Russell小体[13]。免疫组化染色可见GFAP(胶质纤维酸性蛋白,提示胶质细胞来源)呈弥漫性免疫阳性。Vimentin(+)、部分病例CD34(+),EMA弱阳性或阴性,Brachyury (-) 31512680。值得注意的是,脊索样胶质瘤常表现出较低的Ki67增殖指数(5%),表明其生物学行为相对良性[14]。
脊索样胶质瘤具有明显的脊索样表现,具有特征性伴簇状嗜伊红的肿瘤细胞在蓝色黏液基质中分布的组织学特点,但仍需与累积鞍区、第三脑室和侧脑室的肿瘤相鉴别[15][16]。
1.颅咽管瘤:颅咽管瘤是鞍区最常见肿瘤,发病年龄呈10~20岁50岁双峰双峰分布,其起源于拉克氏上皮,多位于鞍上或第三脑室。颅咽管瘤多有囊变钙化,因而T2WI多较混杂,实性部分常明显结节状强化。免疫组化GFAP(-),有利于与脊索样胶质瘤鉴别。
2.垂体大腺瘤:位于鞍区,可见蝶鞍扩大,当肿瘤向上突破鞍膈呈典型“束腰征”,增强扫描病灶均匀强化,T2WI以等信号为主。垂体腺瘤常表现为垂体功能障碍。
3.毛细胞型星形细胞瘤;多见于儿童,可生长于鞍区,较大时可突入三脑室底部,多呈囊实性,壁结节样强化或环形强化。
4.生殖细胞瘤:多位于松果体区,异位生长的生殖细胞瘤可位于鞍区或鞍上,信号多呈稍长T1、稍长T2信号,钙化坏死少见,可有小范围囊变。松果体区若亦有病灶或HCG(人绒毛膜促性腺激素)明显升高有利于诊断。
5.室管膜瘤:多位于侧脑室、四脑室,三脑室室管膜瘤少见,少部分可位于脑内,肿块与脑室壁呈广基底相连,T1WI呈等或低信号,T2WI呈不均匀高信号,肿瘤内可见囊变、钙化或出血,增强后呈不均匀强化。
6.脑膜瘤:脑室内脑膜瘤好发于侧脑室三角区,T1WI呈等或稍低信号,T2WI呈等或稍高信号,增强呈明显均匀强化,囊变坏死少见。脑膜尾征有助于鞍区脑膜瘤的诊断。
7.脉络丛乳头状瘤:好发于儿童,多见于侧脑室三角区,表面呈颗粒状,T1WI呈等或低信号,T2WI呈等或稍高信号,增强后显著强化,多伴有脑积水。
8.脊索瘤:脊索瘤由脊索残余发生,多见于脑中线鞍背和斜坡部,与颅骨关系密切,镜下可见典型的黏液样基质和液滴样细胞,无淋巴、浆细胞浸润,免疫组化GFAP、CEA和D2-40阴性,Brachyury、EMA、CK、S-100和vimentin阳性,Brachyury阳性有助于与脊索样胶质瘤鉴别。
手术切除是脊索样胶质瘤的首选治疗方法。应根据个体肿瘤生长模式进行手术选择。Huo等人报道了一例使用内窥镜辅助经终板入路成功治疗大体全切除的脊索样胶质瘤病例,通过回顾文献中的相关病例,他们提出此入路可能与死亡率显著降低有关[17]。不完全切除与复发率显着增加有关。由于肿瘤靠近甚至粘附于关键神经结构(如下丘脑、视神经和脑脊液循环通路),手术具有重大风险。常见的并发症是视力恶化、垂体困难和梗阻性脑积水[18]此外,尚有患者术后发生可逆性高钠血症或中枢性尿崩症。 若患者发展为阻塞性脑积水,需要脑室-腹膜分流[16]。化疗或放疗作为脊索样胶质瘤辅助治疗的作用尚不清楚[19,20]。尽管脊索样胶质瘤常表现出较低的Ki67增殖指数,但仍具有复发和转移倾向。 Jung等报道了第三脑室脊索样胶质瘤,伴有肿瘤复发和加重的脑积水,其Ki-67标记指数低于1%[21];Ki等人报告了一名脑室内播散且Ki-67标记指数低于0.5%的患者[22];Erwood等人报告了一例次全切除术后快速进展的脊索样胶质瘤病例,尽管其Ki-67增殖指数仅为3~4%[23]。因此,尽管脊索样胶质瘤的组织学性质低,我们仍建议对所有脊索样胶质瘤患者进行长时间的连续观察。
综上所述,尽管鞍上第三脑室脊索样胶质瘤并不常见,患者临床症状可能也不具备特征性表现,通过仔细询问病史,对患者进行全面的体格检查和相应的辅助检查,并仔细分析患者影像资料,可有助于减少漏诊和误诊。通过纵裂胼胝体入路对这类脊索样胶质瘤予以全切可能会出现电解质紊乱。术后需注意密切监测患者水电解质和激素水平,早期发现,及时纠正,积极干预有助于患者的恢复,最终有助于改善患者的神经功能和预后。
参考文献
往期回顾

声明:脑医汇旗下神外资讯、神介资讯、神内资讯、脑医咨询、Ai Brain 所发表内容之知识产权为脑医汇及主办方、原作者等相关权利人所有。
