



在阿尔茨海默病 (Alzheimer'sdisease, AD) 早期,周细胞收缩毛细血管,增加其液压阻力并导致免疫细胞滞留,从而减少脑血流量(CBF)。然而,目前缺乏减轻AD患者周细胞介导的收缩的治疗方法。近期,英国伦敦大学学院DavidAttwell研究小组在《Nature Neuroscience》期刊发表了题为“Inhibiting Ca2+ channelsin Alzheimer’s disease model mice relaxes pericytes, improves cerebral blood flow and reduces immune cell stalling and hypoxia” 的文章,该团队利用激光多普勒、散斑流量计和磁共振成像的体内双光子成像,发现Ca2+通过L型电压门控钙通道(CaV)进入控制周细胞的收缩张力。在AD模型小鼠中,发现整个毛细血管床的周细胞可以诱发免疫活性氧(ROS),同时也是周细胞内钙浓度([Ca2+]i)介导的微血管流量减少的关键驱动因素。在疾病进展的早期用尼莫地平阻断钙通道可改善脑血流量,减少白细胞在周细胞胞体的停滞,减轻脑缺氧。另外,钙通道阻滞也大大减少了人脑皮质组织中淀粉样蛋白β(Aβ)引起的周细胞收缩。因此,在AD早期降低周细胞内的钙浓度可能提供一种增强AD大脑能量供应和认知功能的治疗策略。

周细胞收缩张力由Ca2+通过L型CaV和Ca2+门控TMEM16ACl−通道之间的相互作用来控制。作者通过防止周细胞中的[Ca2+]i升高来减少AD早期发生的CBF降低。Gq蛋白偶联受体(GqPCR)激动剂,如ET-1(AD12中Aβ诱导的细胞外基质收缩的驱动因素),从肌浆网释放Ca2+。这种Ca2+可能直接激活肌动球蛋白或通过TMEM16A通道触发Cl−外流,导致膜去极化和CaV激活,引起Ca2+内流和收缩。对表达tdTomato和钙指示剂GCaMP5g(NG2-CreERT2-GCaMP5g小鼠)的急性皮质切片进行了双光子成像,ET-1增加了细胞外基质[Ca2+]i并收缩了周细胞体的毛细血管(S1b-d),其中大多数介导收缩的细胞外基质过程都存在(图S2a和视频S1)。使用尼莫地平阻断CaVs或使用10bm阻断TMEM16A,显著减弱了[Ca2+]i的升高和周细胞收缩(图S1b-d),并在SMCs中检测到尼莫地平引起的[Ca2+]i升高相似降低,这与CaV1.2(图S3)和TMEM16A在壁细胞中高度表达一致。另外用于溶解尼莫地平的溶剂没有调节周细胞的[Ca2+]i的作用(图S1f)。本文所提出的关于TMEM16A和CaVs如何控制周细胞张力的模型在(图S1g)中展示。
为了评估CaVs是否在体内产生收缩张力,作者使用激光多普勒血流仪测量了脑血管的CBF,并在戊巴比妥麻醉的NG2-dsRed小鼠体内进行了双光子成像,这些小鼠的周细胞和平滑肌细胞被标记为dsRed21(图1a,b)。使用FITC-dextran来可视化血流,在成熟的P120–P158野生型(WT)小鼠的股静脉中注入BBB渗透性CaV阻断剂尼莫地平引起了CBF的长期增加。单独注入溶剂没有改变CBF(图1c),这意味着尼莫地平而不是溶剂或血液体积的小幅度增加提高了CBF。
双光子成像显示,FITC-葡聚糖(图1b)的内腔成像表明,尼莫地诱导的CBF升高与脑膜动脉、穿通动脉和第1-3级毛细血管直径的长期增加相一致(图1d;图S1h,i)。毛细血管扩张到尼莫地平的最大值靠近周细胞体,并且随着与细胞体的距离增加而显著降低(图1e),这与周细胞介导尼莫地平诱导的毛细血管扩张一致。同时测量CBF和血压(BP)显示,静脉注射尼莫地平诱导了平均血压的初始下降(可能是由于周围血管扩张)从注射后7.5分钟的88mmHg下降到67mmHg(图1f),持续约30分钟。这与CBF的小幅初始下降相关,但随着自身调节机制将血压恢复到原始值,CBF升高到大约比尼莫地平前值高25%。这些数据表明,尼莫地平引起的局部血管阻力降低大于其对全身血压的影响(也有报道另一种CaV阻断剂可引起血压降低同时增加脑血流量)。
因为Ca2+通过CaVs流入导致周细胞收缩(图S1b-g),推测壁细胞[Ca2+]i的降低将先于尼莫地平诱导的CBF增加。通过激光多普勒流速计和双光子成像技术检测NG2-CreERT2-GCaMP5g小鼠血管直径和壁细胞[Ca2+]i,作者证实了尼莫地平诱导的[Ca2+]i在CBF增加之前降低(第一至第三级周细胞体14%,第一至第三级周细胞突起32%,平滑肌细胞22%),并与血管扩张相关(图1g,h,j):周细胞体[Ca2+]i峰值降低一半的平均时间和CBF峰值增加一半的平均时间有显著差异(图1i)。因此,SMCs和第一至第三级周细胞中的CaVs在WT小鼠中产生了肌源性张力。
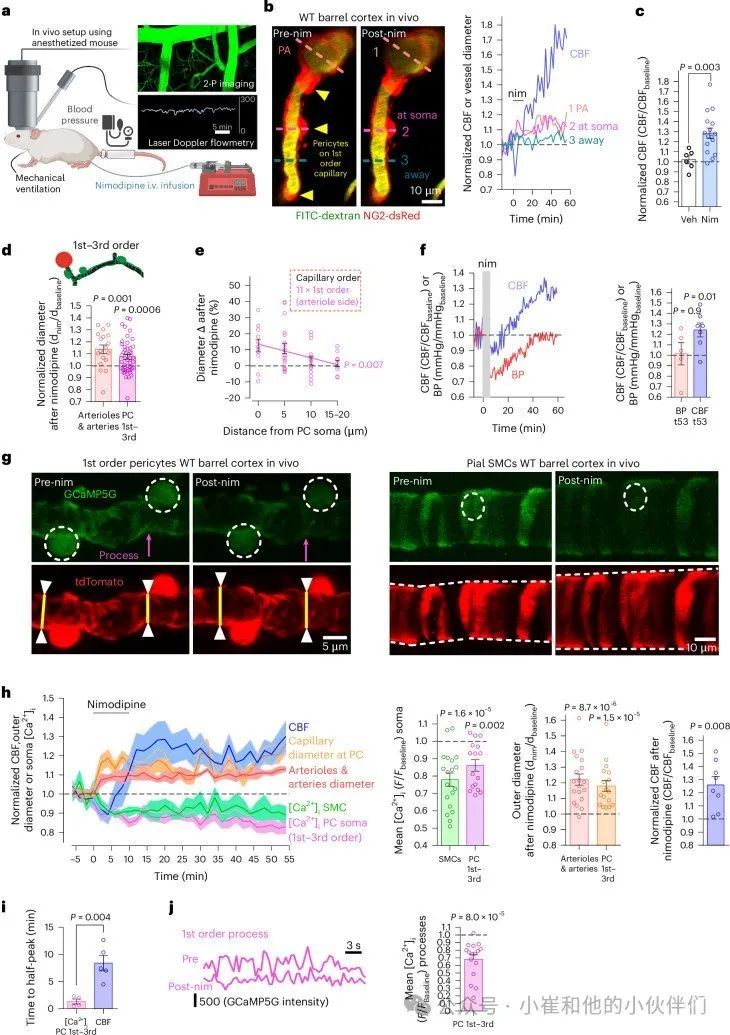
图1 CaVs在WT小鼠体内的SMCs和第一至第三级血管周细胞中产生张力
结果二:CaVs增强毛细血管床的周细胞收缩
CaVs增强毛细血管床的周细胞收缩,人类AD患者早期CBF下降,与周细胞介导的毛细血管收缩一致(在小鼠AD模型中,毛细血管收缩,但动脉和静脉不收缩)。因此,作者测试了在NG2-dsRedxAPPNL-G-F小鼠模型中CaVs是否参与了周细胞介导的毛细血管收缩,该模型敲入了瑞典、北极和伊比利亚APP突变以避免APP过表达产生伪影,并且壁细胞用dsRed标记,并将APPNL–G–F小鼠命名为AD小鼠。与其他AD模型中的CBF下降一致,通过激光散斑成像,发现与野生型小鼠相比,P191-P206AD小鼠的大脑皮层毛细血管灌注显著减少(图2a)。在成熟的P125-P158AD小鼠中,体内双光子成像结果的第一至第三级毛细血管显示,与同龄P120-P158野生型小鼠相比,周细胞体的毛细血管腔狭窄了21%(图2b)。类似的小血管收缩在其他AD小鼠模型中也可见,可能由Aβ诱导的ROS和ET-1释放收缩周细胞驱动。
尼莫地平治疗成熟AD小鼠后,1-3级周细胞体毛细血管直径增加了9.6%至6μm,即接近1-3级WT小鼠毛细血管直径的6.9μm(图2b),并使CBF增加51%(图2c)。为了测试尼莫地平诱导的血管舒张是否通过降低壁细胞[Ca2+]i来增加CBF,作者在成熟ADxNG2-CreERT2-GCaMP5g小鼠中结合了激光多普勒流速仪和双光子成像。与WT小鼠(图1h)一样,在这些AD小鼠中,尼莫地平诱导的壁细胞体[Ca2+]i下降与毛细血管和动脉直径的增加以及CBF的35%缓慢上升(图2d)相关:周细胞体[Ca2+]i峰值下降一半的平均时间和CBF峰值增加一半的平均时间差异显著(图2e)。尼莫地平显著降低了AD小鼠重的1-3级周细胞过程的平均[Ca2+]i值(图2f),其覆盖的毛细血管程度与WT小鼠相似。
对于图2d中同时记录的直径和血流时间序列数据,在使用尼莫地平后,周细胞处的动脉和毛细血管的外径分别增加了17.8%和16.9%(这略微低估了管腔直径的变化),CPF增加了约40%。为评估尼莫地平引起的毛细血管和动脉直径增加是否可以解释观察到的CBF增加,作者使用了先前的一个数学模型,其中43%的血管床阻力在动脉(阻力与直径的4次方成反比)和57%在毛细血管(阻力取决于周细胞周围直径的空间分布)。在AD引起的周细胞处毛细血管直径21%的降低(图2b)以及动脉或静脉直径无变化之后,该模型预测尼莫地平引起的在周细胞处的毛细血管直径增加16.9%和动脉直径增加17.8%(图2d),将使CBF增加51%,与激光多普勒测量观察到的40-51%增加相似(图2c,d)。可以得出结论,尼莫地平对CBF的影响是通过它引起的血管直径增加来解释的,无需调用尼莫地平对其他细胞(如周细胞和SMCs)的影响。在1-3级分支的周细胞中,AD小鼠的[Ca2+]i瞬时频率在细胞体中增加了3.7倍,在突起中增加了2.6倍(图2g,h),而周细胞突起的[Ca2+]i瞬时幅度(图2g)或SMC[Ca2+]i瞬时频率或幅度(图2i,j)没有显著变化。用尼莫地平治疗AD小鼠在很大程度上将1-3级周细胞突起的[Ca2+]i瞬时频率恢复至野生型水平(图2g)并降低了瞬时幅度(图2g)。同时,溶剂处理没有显著调节SMC或周细胞的[Ca2+]i,也没有调节动脉、小动脉或毛细血管的直径(图2d)。重复使用800nm光(对GCaMP5G荧光缺乏Ca2+敏感性)成像引起恒定的荧光,没有可见的瞬态或光漂白衰减(图2Se-g和视频S3),这表明光漂白、运动伪影和制备下降没有影响测量的[Ca2+]i。尼莫地平因此降低了1-3级周细胞的波动以及平均[Ca2+]i。比较19只成熟WT小鼠和11只成熟AD小鼠(P110-P191),在所有基因型(无论dsRed(图2c)或NG2-CreERT2-GCaMP5g(图2d)共表达)中尼莫地平引起的CBF均增加,AD小鼠的CBF增加比WT小鼠大74%(上升47%),而WT小鼠的CBF增加为27%,这表明AD小鼠中CaV引起的紧张度更大(正如预期的那样,周细胞介导的毛细血管收缩与AD12中的CaVs产生的周细胞紧张度相关)。因此,在AD中阻断CaVs,例如使用尼莫地平,是逆转AD早期发生的CBF下降的潜在途径(图2k)。
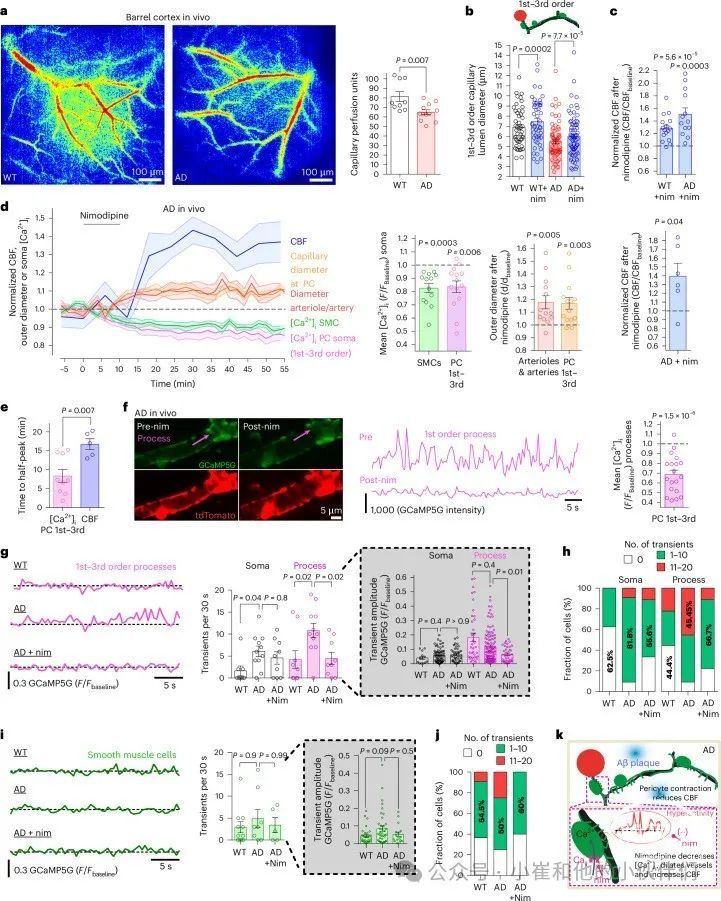
图2 周细胞介导的毛细血管收缩和CBF降低与CaVs有关
尽管1-3级毛细血管上的收缩周细胞可以快速调节毛细管直径,但作者发现毛细管床中部和小静脉侧的毛细血管(以下简称“>3级”),占WT小鼠总毛细血管长度的91%,占AD小鼠的94%(图S4a)。因为>3级周细胞可能会在缓慢的时间尺度上产生收缩张力并占据大部分毛细血管床(大脑大部分血管阻力所在的地方),因此它们可能会对CBF产生强烈影响从而响应尼莫地平。然而,在体内测量WT小鼠周细胞体的>3级毛细管直径显示,静脉注射尼莫地平处理后毛细管直径没有显着变化(图3a、b)。在WT小鼠中注射尼莫地平后的小静脉和静脉的直径也没有显着变化。相比之下,在AD小鼠中,与WT小鼠相比,>3级毛细血管在周细胞体细胞处明显更加收缩(13.5%),并且静脉注射尼莫地平治疗部分逆转了这种收缩(图3b),这与尼莫地平减少>3级周细胞体细胞和过程中的[Ca2+]i一致(图3c)。值得注意的是,尽管与WT小鼠相比,AD的>3级周细胞体细胞中的[Ca2+]i瞬变频率升高,但尼莫地平并不调节>3级周细胞的胞体或过程中的[Ca2+]i瞬变频率,但它确实降低了瞬变的幅度(图S2h)。>3级周细胞响应缺血损伤调整毛细血管直径的能力一直存在争议。为了测试>3级周细胞是否可以收缩(可能发生在缺血和AD皮层中,其中血管收缩剂(如ROS)水平升高),诱导了产生ROS的双光子激光实质损伤,激光束瞄准距离NG2-CreERT2-GCaMP5gWT小鼠皮层中单个毛细血管约15-20μm的距离(图3d)。在3级或更高级别的周细胞中检测到强烈的[Ca2+]i升高,这与周细胞胞体的毛细血管收缩相吻合,但在毛细血管腔部分≥距离这些胞体20μm处的收缩显着减少(图3e-g,图S4b和视频S4)。激光诱发的周细胞收缩也强烈增强了血液的停滞,在充满染料的血浆中表现为阴影(图3h)。令人惊讶的是,在1-2级毛细血管或小动脉附近相同的激光诱发损伤并不调节周细胞或SMC中的血管直径或[Ca2+]i(图S4b-d和视频S5)。尽管壁画细胞对局部激光诱发损伤的反应因此沿维管树不同,但这些数据证实>3级周细胞可以产生毛细血管收缩。
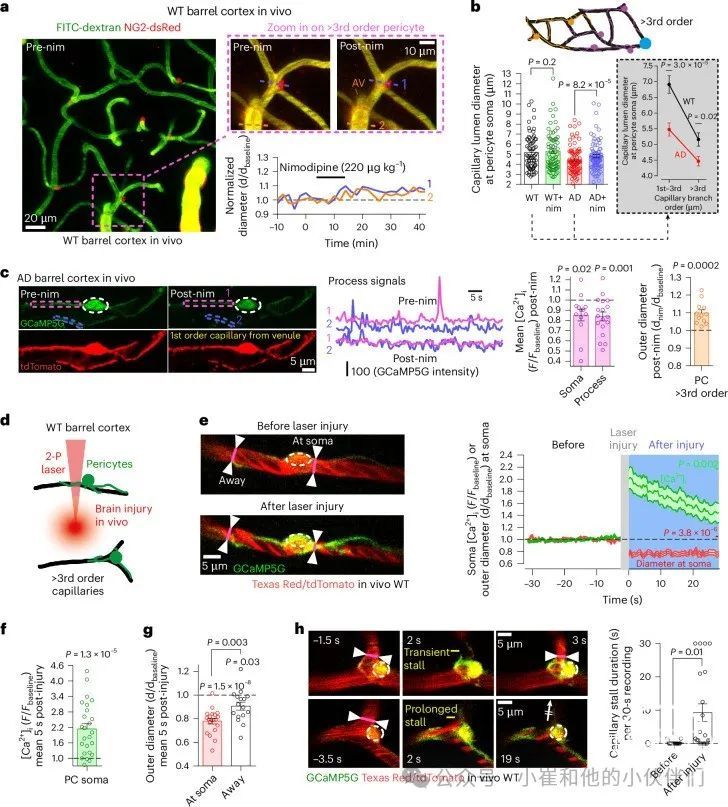
图3 AD小鼠中的中内皮床周细胞表现出增强的收缩张力
结果三:AD小鼠中的免疫细胞ROS导致[Ca2+]i升高并降低CBF
上述数据显示,在AD皮层活体中,周细胞CaVs介导整个毛细血管床的毛细血管收缩,但收缩机制尚不清楚。当Aβ寡聚体急性应用于WT脑切片时,会产生ROS,释放ET-1,从而通过激活CaV通道引发收缩。同样,氧化应激在人类AD37和AD模型中发生,损害CBF。在NG2-CreERT2-GCaMP5g小鼠中进行活体双光子成像(图4a),作者发现通过将1 mM H2O2局部应用于暴露的桶状皮层,产生氧化应激,可以使周细胞[Ca2+]i升高2.07倍(图4a),与ROS引发周细胞收缩的报道一致。H2O2引发的[Ca2+]i升高随皮层深度降低(图4a),表明H2O2扩散限于皮层上层。对于匹配皮层深度的周细胞,尼莫地平将H2O2引发的[Ca2+]i升高降低54%至1.49倍(图4a),因此CaVs至少介导了活体中一半的[Ca2+]i升高。尼莫地平还大大降低了SMCs中的H2O2引发的[Ca2+]i升高。
微血管周细胞收缩由急性应用Aβ引起,部分依赖于NOX2介导的小胶质细胞和血管周围巨噬细胞(PVM)产生的活性氧(ROS),在大脑中,包括在人类阿尔茨海默病中,这些细胞表达在小胶质细胞和PVMs中(图S5a和S6g)。小胶质细胞和PVMs的贡献可能因毛细血管床的位置而异,因为PVMs在动脉和毛细血管床的前几个分支周围更多(图S6c,d)。为了测试Aβ在阿尔茨海默病中积累是否同样会在小胶质细胞中产生ROS,作者将野生型(WT)和阿尔茨海默病(AD)小鼠的急性皮质切片与活性氧传感器二氢乙锭(DHE)共同孵育,当氧化DHE嵌入DNA时,DHE会发出荧光。在异硫氰酸荧光素B4标记的小胶质细胞/PVMs中,与野生型小鼠相比,AD小鼠的DHE荧光(归一化到无DHE时的自荧光)增加了49%(图4c,d),尽管AD小鼠的自荧光更高。同样,使用活性氧传感器CellROX检测表达增强型绿色荧光蛋白(eGFP)的ADxNG2-dsRed小鼠皮质切片中证实了AD中小胶质细胞/PVMs的ROS产生(图S5k)。相比之下,在周细胞中也没有检测到DHE荧光的变化,在其他血管细胞(不是IB4标记的周细胞)或除小胶质细胞/PVMs以外的血管外细胞中也没有检测到(定义为IB4阴性细胞,在IB4标记的皮层切片的血管外空间中呈阴影状(图S5b)。此外,通过MitoSOX测量野生型小鼠和AD小鼠的微胶质细胞或周细胞中的线粒体ROS,结果显示两者之间没有差异(图S5c)。
ROS水平的升高可能通过触发周细胞CaVs的Ca2+流入导致AD小鼠CBF降低(图4a,b),因此作者测试了通过阻断 NOX2 介导的微胶质细胞/PVM 产生 ROS 是否可以调节 AD 小鼠的 CBF。将NOX2阻断剂GSK2795039(GSK)局部应用于AD小鼠的皮层表面,与安慰剂治疗相比,体内的CBF增加了42%(图4e)。在其他AD小鼠模型中,也报道了使用其他NOX2阻断剂有类似的毛细血管血流改善。为了更好地模拟临床情况,因此作者测试了临床批准的ROS清除剂、BBB渗透性抗氧化剂N-乙酰半胱氨酸(NAC)如何调节AD小鼠的CBF。NAC使脑血流量增加62%(图4f)并降低周细胞和平滑肌细胞中的[Ca2+]i(图4g),与NAC降低周细胞中ROS诱导的[Ca2+]i升高和阻断其收缩的结果一致。与抗氧化剂谷胱甘肽治疗相比,与NAC不同,谷胱甘肽难以穿过血脑屏障(因此,主要清除周围活性氧),并未调节脑血流量(图S5d)。
因为Aβ斑块是Aβ寡聚体和活性氧(ROS)的储存库,驱动小胶质细胞产生独特的炎症表型,作者研究了ROS的产生与小胶质细胞中在靠近(距Aβ斑块<65 μm)与远离(距Aβ斑块>65 μm)斑块时是否不同,以及ROS是否可以被NAC靶向。使用ROS传感器(CellROX),作者测量了ADxNG2-dsRedxIba1-eGFP小鼠皮层切片中的ROS。组别分别是:(1)斑块中表达Iba-1的小胶质细胞,(2)远离斑块的表达Iba1的小胶质细胞和(3)与斑块相关的小胶质细胞,这些细胞同时表达Iba1和P2Y12受体(常用作小胶质细胞标记)以及血管周细胞和少突胶质细胞前体细胞(OPC)标记的NG2(图S5e–h)。这个第三类细胞在面神经轴突切断、缺血、帕金森病和AD中曾被注意到,转录组特征分析表明这些细胞具有高度的增殖性。NG2 表达的小胶质细胞主要分布在斑块处(图S5f,i),在AD皮层中随年龄增长数量增加(斑块负担随年龄加重),但在WT小鼠大脑中不存在,并且仅在AD小脑中晚期出现(图S5j),而在AD小脑中Aβ沉积出现得非常晚,且该区域的CBF无变化。添加CellROX后,在Iba1标记的小胶质细胞中,皮层中的荧光在633 nm波长下增加了57%(在斑块处),在斑块外增加了45%,在NG2表达的小胶质细胞中增加了33%(图S5k)。预先用50 μM NAC孵育,小胶质细胞中的CellROX引起的荧光在斑块处和斑块外分别增加至16%和18%,但并未显著降低表达NG2的小胶质细胞的CellROX荧光(图S5k)。因此,NAC在阿尔茨海默病小鼠中在很大程度上阻止了ROS的产生。
与阿尔茨海默病和脑炎症期间小胶质细胞向血管迁移结果一致,作者发现与WT小鼠相比,在活体小脑皮层和ADxNG2-dsRedxIba-eGFP小鼠的皮质中,Iba1标记的小胶质细胞/PVM体和周细胞体的距离减小(图4h和图S6a,b)。这在第一至第三级和第三级以上分支的毛细血管上的周细胞都是如此(图S6b),因此,在AD小鼠的毛细血管床中,小胶质细胞/PVM细胞体与周细胞细胞体更接近。在4-6个月大的WT和AD小鼠中,每根毛细血管长度的周细胞密度(图S2b)、小胶质细胞密度(图4i)、PVM密度(图S6c,d)和脑萎缩(见下文)相同,排除了这些参数变化导致AD小鼠中周细胞与小胶质细胞更接近的可能性。有趣的是,据报道,在人类AD中,位于额上回的小胶质细胞与周细胞小于10 μm的比例降低,但在作者的小鼠中,AD中位于周细胞细胞体10 μm范围内的微胶质细胞体比例增加(图S6a)。在人类研究中,只有大约4%的小胶质细胞与周细胞的距离小于10 μm,因此周细胞和微胶质细胞平均距离将由96%的未“与周细胞相关”的微胶质细胞主导。这些差异的原因与人类研究不确定,但它们可能反映了人类具有更先进的AD(比作者的小鼠),这与周细胞丢失有关(与作者的研究不同:图S2b),或者研究的是不同的脑区。与AD中Iba1表达细胞与周细胞的增强相互作用一致(图4h),作者发现与WT小鼠相比,ADxNG2-dsRed小鼠中P2Y12R标记的小胶质细胞体或CD206标记的PVM细胞体与第一至第三分支级周细胞体之间的距离分别减少了27%和67%(图S6f)。
在WT和ADxIba1-eGFP小鼠的桶状皮层进行体内成像显示,与WT小鼠相比,AD小鼠中微胶质细胞累积像素监控(超过20分钟)显著减少(图4j,k)。然而,在AD小鼠中与细胞大小无关的运动(每个细胞的监视指数除以其面积)没有变化(图4l),这表明减少的监控反映了AD小鼠比WT小鼠中的微胶质细胞分支更少(图4j)。因此,尽管AD中监控的减少可能会导致小胶质与毛细血管的相互作用过程减少,但在AD小鼠中,小胶质/PVM细胞体与血管的距离比WT小鼠更近,这可能会缩短释放的ROS需要扩散以调节血管张力的距离。图4m总结了受损的血管和免疫细胞相互作用如何引发AD中的周细胞收缩的过程。
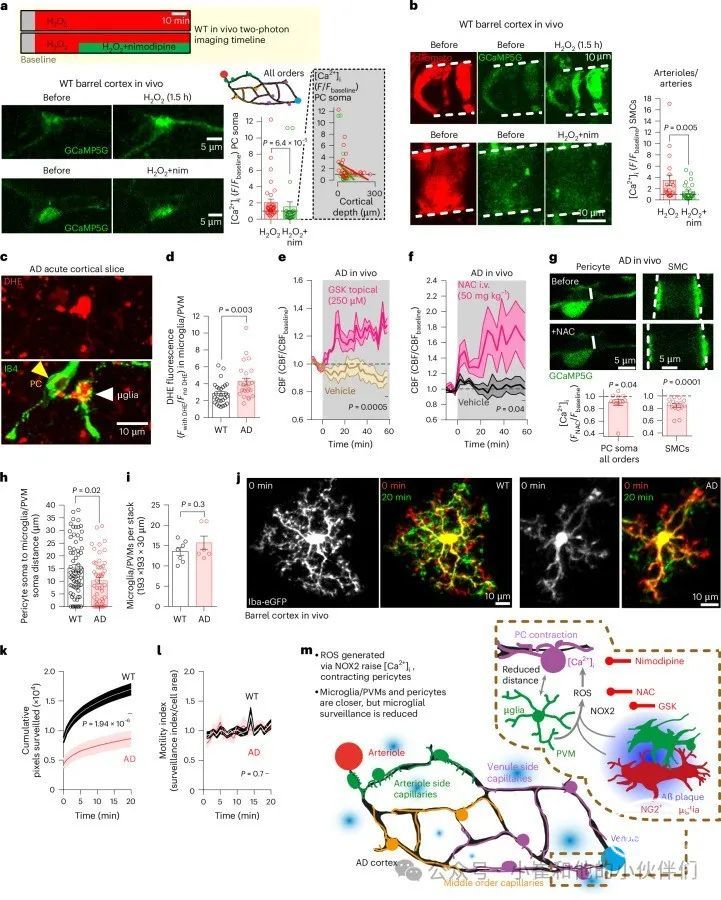
图4 来自大脑免疫细胞的ROS驱动AD小鼠的周细胞收缩
结果四:AD小鼠中的小静脉末梢周细胞驱动白细胞停滞
微血管周细胞驱动AD小鼠中的白细胞停滞。为测试AD小鼠中周细胞引起的毛细血管腔狭窄(图2b,d和3b,c)是否会导致血液在周细胞体附近停滞,作者获得了约3-5秒(取决于毛细血管方向,约3-5帧)的三维体内双光子成像堆栈,以捕获每个毛细血管段。如先前报道所述,作者发现阻塞毛细血管的百分比从WT小鼠的0.47%增加到AD小鼠的5.2%(图5a和视频S6)。如果毛细血管中的块状物随机分布,那么块状物出现的概率应在上皮细胞间距的一半距离内保持恒定,这对于每个144 μm有一个上皮细胞的毛细血管(图S2b)来说,是2 μm(在更大的距离上,块状物将更接近沿毛细血管的下一个上皮细胞)。测量从毛细血管中119个区块位点到最近血管周细胞体的距离,发现随机区块位置的累积概率分布(在72 μm处达到1的直线)与实验观察到的分布显著不同;区块与血管周细胞之间的距离比随机定位的毛细血管区块更近(图5b)。
从AD小鼠的肺动脉(PAs)追踪毛细血管分支顺序,1-2级毛细血管中没有区块,3级毛细血管中有13%,从PAs的≥4级毛细血管中有87%。从升静脉(AVs)追踪分支顺序,87%的区块位于AVs的1-3级毛细血管分支中(图5c)。因此,毛细血管阻塞主要发生在毛细血管床的静脉端,此处毛细血管直径最小(在AD中进一步减小,图3b)。
因为循环的单核细胞和中性粒细胞被招募到AD脑中,它们的可扩张性较低且比红细胞大,作者通过使用ADxIba1-eGFP小鼠成像单核细胞和使用针对Ly6G的低剂量荧光偶联抗体(0.1mg kg−1i.v.,不会引起粒细胞减少)来研究中性粒细胞对毛细血管堵塞的贡献。在三维成像堆栈中(图5a),作者发现1.7%的毛细血管显示出含有中性粒细胞的堵塞,0.9%显示出含有单核细胞的堵塞(图5d)。作者没有同时标记中性粒细胞和单核细胞(因此含有中性粒细胞的堵塞也可能含有单核细胞),但共同来看,这些可能解释了5.2%的毛细血管堵塞的一半(图5a),其余的堵塞可能含有红细胞(见下文)。由于白细胞堵塞持续时间决定了堵塞引起的CBF降低的程度,作者通过在单平面中以2Hz的频率成像毛细血管来捕捉堵塞的开始和结束(视频S7)。中性粒细胞的平均堵塞持续时间比单核细胞长5.4倍(图5e),可能是因为它们的灵活性较低或更倾向于附着在内皮细胞上(一些单核细胞也暂时附着于硬脑膜静脉或动脉,但并未阻止血流(图5d和视频S8))。
与由周细胞引起的毛细血管狭窄导致白细胞停滞一致,作者发现中性粒细胞停滞处的毛细血管直径明显小于中性粒细胞直径,导致中性粒细胞推压毛细血管壁(图5f和视频S9),这可能会增强它们与内皮细胞上的ICAM-1和VCAM-1粘附分子的相互作用(图S7a-f)。事实上,低剂量的抗Ly6G,它结合Ly6G并因此减少中性粒细胞表面整合素的表达,从而减少整合素与ICAM-1的结合(而不引起中性粒细胞死亡),增加了AD小鼠的脑血流量,但未增加WT小鼠的脑血流量(图S7j,k)。与中性粒细胞不同,停滞的单核细胞直径并不大于它们停滞的毛细血管段直径(图5g)。
如果由周细胞引起的毛细血管收缩导致白细胞停滞,那么使用尼莫地平增加毛细血管直径应该可以缓解毛细血管阻塞。作者使用FITC-白蛋白灌注方案来可视化固定皮层切片中所有在体内保持通畅的血管,以及有助于阻塞非通畅毛细血管的附着细胞(方法)。在给予载体的AD小鼠中,作者检测到与WT小鼠相比,靠近(但不是远离)Aβ斑块附近的毛细血管存在显著的灌注不足(图5h,i)。此外,与WT小鼠相比,AD小鼠的皮层毛细血管中含有更多阻塞的白细胞(CD45标记)、中性粒细胞(Ly6G)和红细胞(ter119)(图5h,j,k)。值得注意的是,在AD小脑(几乎不含Aβ沉积物)中,毛细血管灌注和中性粒细胞阻塞毛细血管未受影响(图S7i)。AD中皮质毛细血管中白细胞停滞的增加并不反映血液白细胞计数更高(图S7g和图S1)。此外,与中性粒细胞相关的阻塞发生在周细胞体附近(图S7h),表明血管周围细胞通过收缩毛细血管来驱动血管。确实,作者发现,在用尼莫地平在体内治疗1.5小时的AD小鼠中,毛细血管灌注得到了很大程度的恢复,停滞的白细胞和红细胞数量减少到低于WT计数(图5i-k)。因此,尼莫地平通过几种方式增加AD中的CBF:通过逆转发生的毛细血管直径减少(从而也保持血液粘度较低)以及通过减少白细胞和红细胞导致的毛细血管阻塞。
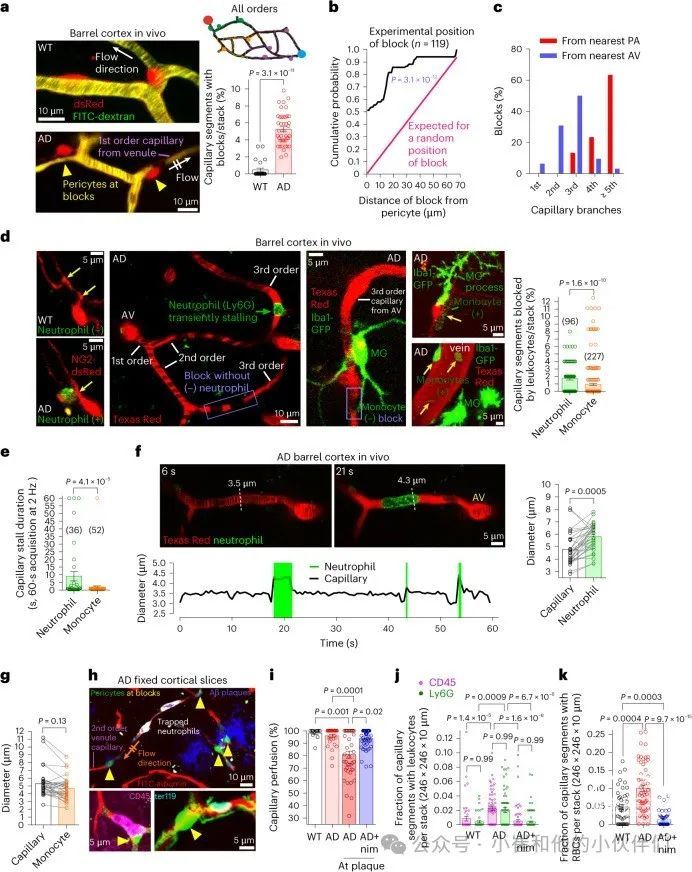
图5 抑制CaV减少了AD中由周细胞引发的血细胞引起的微血管阻塞
结果五:尼莫地平改善AD小鼠的CBF并减少脑缺氧
建立了尼莫地平逆转AD小鼠CBF减少的方法后,为了模拟早期AD临床治疗情景(假设早期AD诊断),作者用尼莫地平或溶剂处理AD小鼠饮用水1.5个月(图6a和图S8a,b),从2-3个月大的年龄开始,这个年龄标志着突触丢失和Aβ斑块沉积的开始,但远在记忆丧失之前。使用体内双光子成像,作者发现尼莫地平治疗显著增加了皮质血管中的毛细血管直径(图6b)并大量减少了毛细血管中的血液停滞(图6c和视频S10)。在溶剂处理或尼莫地平处理的1-3级毛细血管中,停滞现象基本不存在(图5c和6c)。为了测试尼莫地平是否也能调节皮质下区域的CBF(无法通过双光子显微镜、激光多普勒流速仪或激光散斑成像获得),作者使用了动脉自旋标记(ASL)MRI来非侵入性地测量丘脑的CBF。结果表明长期使用尼莫地平治疗显著增加了AD小鼠的丘脑CBF(图6d)。
使用匹莫尼达唑后在体内测量脑缺氧显示,AD增加了靠近Aβ斑块的大脑皮层和海马中的缺氧细胞数量,而在尼莫地平治疗1.5个月后,缺氧细胞数量显著减少(图6e、f)。尼莫地平还显着降低了溶酶体相关膜蛋白1(LAMP1)标记的斑块相关溶酶体(在小胶质细胞和轴突肿胀中)的数量(图6g)。这些由尼莫地平引起的改变不太可能是由于大脑萎缩的变化所引起的,因为皮质和海马区域在给予溶剂和尼莫地平治疗的鼠中相似(图S8c)。1.5个月的尼莫地平治疗没有调节毛细血管、小动脉或小静脉中内皮细胞ICAM-1或VCAM-1的表达,也没有调节在三个给予溶剂和四个尼莫地平治疗的成熟AD小鼠中FITC-葡聚糖的渗漏(P=0.6和P=0.8,来自Mann-Whitney检验,对于皮质中深度小于40 μm和40-126 μm处的外渗分别进行测量)。与野生型小鼠相比,未经治疗的AD小鼠的FITC-dextran外渗和纤维蛋白原沉积增强(图S9a-e)。对于FITC-dextran,外渗在靠近皮质表面的40 μm区域最大(图S9b,c)。成熟和老年AD小鼠在皮质表面下方40 μm以上处显著增加了外(图S9b)。在AD中,纤维蛋白原沉积最大的是在靠近淀粉样斑块的区域(图S9d,e)。在治疗1.5个月后,在载体组和尼莫地平组之间未检测到水消耗、体重、呼吸频率或Aβ斑块负荷的差异(图8Sa-d)。在另一组接受载体和尼莫地平饮水治疗的AD小鼠中,作者检测到毛细血管直径的增加和毛细血管停滞的减少(图S8e–g),表明尼莫地平在1.5个月的治疗的早期就恢复了CBF,并且这种改善至少持续1.5个月的尼莫地平治疗(图6b)。
由于CBF在衰老的AD大脑中下降,作者试图研究急性尼莫地平治疗对老年WT和AD小鼠CBF的影响。使用激光多普勒血流仪,作者发现与成熟的(P110–P200)AD小鼠相比,尼莫地平引起的CBF增加在老年(>P200)AD小鼠中显著减弱(减少63%,P=0.03)(图S10a)。尽管在WT中,尼莫地平引起的CBF升高与衰老呈正相关,但在AD中,尼莫地平引起的CBF升高与衰老呈负相关,并且可能与皮质Aβ斑块负荷有关(图S10b–d)。尽管在老年AD小鼠中,动脉、小动脉和1-3级毛细血管对尼莫地平的反应产生了扩张,但尼莫地平引起的>3级毛细血管扩张在治疗1小时内基本消失(图S10e)。在晚期AD中,这加上1:周细胞的加速丧失(图S10f),这将导致血脑屏障破裂(部分原因是白细胞黏附分子的上调),2:壁细胞CaVs的下调(图S3e)以及3:突触和神经元的丢失,这意味着降低壁细胞内钙离子浓度的药物可能为AD的预防或早期治疗提供最大的益处。
为了测试CaVs是否参与人类Aβ诱导的周细胞收缩,作者将活人皮层组织与人工脑脊液(aCSF)单独或与可溶性Aβ寡聚体在无尼莫地平或存在尼莫地平的情况下共同孵育(图6h)。在单独的aCSF中,与周细胞体近处相比,毛细血管外径显著扩张(图6i),如先前报道,可能反映了周细胞体释放诱导内皮管生长的因子。Aβ寡聚体显著收缩了周细胞体近处的毛细血管,但在周细胞体远处没有引起显著的直径减小(图6i)。然而,当在持续存在尼莫地平的情况下应用Aβ寡聚体时,周细胞体处的毛细血管直径与接受载体处理的毛细血管没有显著差异(图6i)。因此,在活的人脑组织中,由Aβ诱导的周细胞收缩是由L型钙通道(CaVs)的激活驱动的,可能是人类周细胞中表达的CaV1.2亚型(图S3)。因此,在患有阿尔茨海默病的人类中,像在老鼠中一样,尼莫地平也会增加毛细血管直径并减少缺氧。
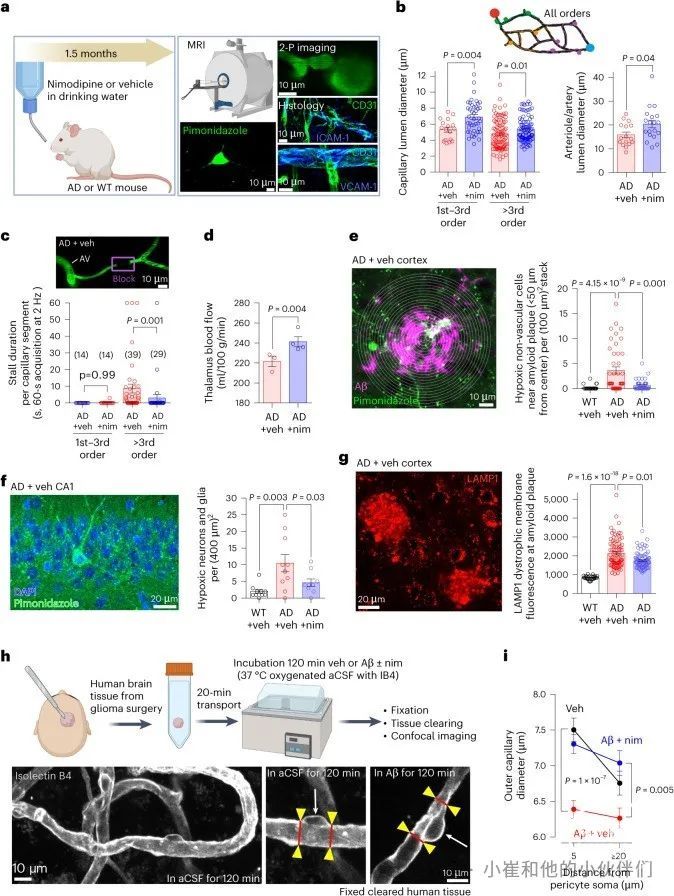
图6 尼莫地平可以改善AD小鼠的CBF并减少脑缺氧,减少人脑组织中Aβ诱导的周细胞收缩
结 论
作者针对AD患者中由周细胞引起的毛细血管收缩的几个机制进行了研究:通过使用血脑屏障穿透阻断剂尼莫地平阻断L型钙通道来调控钙离子流入;通过Ca2+-门控的TMEM16A Cl−通道阻断剂10 bm放大[iCa2+]i的升高;以及使用抗氧化剂(NAC和GSK)的减少活性氧对内皮细胞收缩的作用。这些药物降低了周细胞介导的毛细血管收缩(图2-4和图S1),并增加了CBF。尼莫地平通过放松内皮细胞不仅直接增加了脑血流量,还减少了靠近内皮细胞被捕获的白细胞和红细胞,以及停滞的微血管血流(图5i-k)。因此,尼莫地平可以消除AD大脑中的缺氧(图6e,f)。因此,靶向L型钙通道、TMEM16A或活性氧可能有助于开发维持脑血流量和改善神经功能的治疗方法。
虽然本研究中的大多数机制实验是在麻醉小鼠上进行的,但作者还发现尼莫地平可以扩张清醒未麻醉小鼠的毛细血管和动脉/动脉(图S1h,i);并且给未麻醉的AD小鼠饮用含尼莫地平的水1.5个月后,可以引起周细胞松弛、增加血流、减少毛细血管停滞、组织缺氧和退行性膜标记(图6a-g)。在人类组织中,尼莫地平可以逆转由Aβ诱导的和由周细胞介导的毛细血管收缩(图6h,i),这表明通过使用能够通过血脑屏障的CaV阻断剂可以增加AD患者的脑血流量。
在针对阿尔茨海默病的400多种假想疗法中,大多数未能阻止认知能力下降,可能是因为它们是在不可逆的脑损伤发生后给予的。这表明,针对阿尔茨海默病的未来成功疗法将涉及预防(就像使用降低血压的药物来降低中风风险),或者,如果开发了特定的生物标志物,则治疗疾病的早期阶段。针对阿尔茨海默病早期发生的CBF下降,并在受影响区域达到 45%,足以引起认知变化和神经元细胞损伤,很有吸引力,因为CBF可以通过动态对比增强(DSC)MRI7技术相对无创地测量(并且将来可能使用 ASL MRI 技术)。
在人类患者和小鼠中,毛细血管周细胞在AD早期可以收缩毛细血管。在小鼠中,这种情况发生在小动脉或小静脉直径没有变化的情况下,这意味着AD早期CBF的降低是由周细胞产生的。周细胞介导的毛细血管收缩通过直径减小(通过泊肃叶定律)、较小的直径增加血液粘度以及通过诱导血液细胞引起的毛细血管阻塞来降低CBF。毛细血管阻塞也可能由转基因诱导的周细胞从毛细血管中丢失引起的内皮粘附分子上调,这可能类似于AD晚期发生的周细胞丢失(图S10f)。在 AD 老鼠中,中性粒细胞、单核细胞和红细胞都参与了在周细胞附近产生毛细血管阻塞(图5),从长远来看,这可能导致毛细血管丢失。
作者专注于CaVs,是因为这些通道有临床可用的药物可以针对它们,这些药物可能被重新用于预防或早期治疗AD。一些流行病学分析发现,接受CaV阻滞剂治疗的患者的认知能力下降较少,尽管许多这样的阻滞剂具有较差的血脑屏障通透性,并且它们可能通过降低血压起作用(自调节机制反对这一点,但在AD中可能减弱)。尼莫地平引起的壁细胞[Ca2+]i 下降和体内血管直径增加直接表明尼莫地平可以放松周细胞(在周细胞体附近引起扩张)和动脉周围的平滑肌细胞,这将都会增加脑血流量。据推测,尼莫地平抑制这些细胞中的 Ca2+进入,从而引起扩张(就像在壁细胞中缺失CaV1.2时一样)。考虑到尼莫地平是否也可能作用于神经元以减少触发收缩的ROS的产生,但这似乎不太可能,因为作者没有观察到除小胶质细胞或血管周围巨噬细胞以外的细胞中ROS产生的增加(图4c,d和图S5b,k)。尼莫地平不影响Aβ斑块占据的区域(图S8d),表明它不会改变Aβ的产生,并且NO释放的作用不太可能解释作者的结果:如果尼莫地平作用于内皮或神经元的L型 Ca2+通道,它将降低[Ca2+]i,减少NOS激活和减少NO释放,从而促进收缩而不是扩张。此外,考虑到尼莫地平在中枢神经系统(CNS)中达到的浓度、其作用速度及其对钙通道抑制的半数最大抑制浓度(IC50)或通过其他潜在机制的作用,表明只有钙通道抑制可能有助于增加 CBF(见补充信息)。
APPNL–G–F小鼠为作者提供了一个良好的模型,该模型表现出AD中降低 CBF和升高的Aβ产生,但只有轻微的行为变化,尤其是在4个月大时,作者选择这个年龄是为了防止AD的早期阶段。这突显了确定长期尼莫地平治疗如何影响认知功能的需求。
该概念认为,预防早期AD中由周细胞介导的CBF下降可以显著改善认知衰退,这一观点得到了以下数据的支持:一种防止中性粒细胞在毛细血管中停滞的抗体,它增加了CBF,改善了小鼠晚期的认知。然而,在一项3期临床试验中,一种与尼莫地平相关的药物 尼伐地平,它也增加了CBF,并没有减少轻度至中度AD的人类患者的认知衰退,可能是因为该药物仅在症状出现后4.3年开始使用。此外,尽管降低CBF是AD中Aβ生成的早期结果,但Aβ还有其他作用,包括抑制谷氨酸摄取(这改变了神经兴奋性)和促进tau超磷酸化(这破坏了突触功能)。因此,针对Aβ寡聚体产生和tau磷酸化的疗法可能与旨在维持CBF的疗法协同作用(如尼莫地平)。
精读原文请下载:
Korte N, Barkaway A, Wells J, Freitas F, Sethi H, Andrews SP, Skidmore J, Stevens B, Attwell D. Inhibiting Ca (2+) channels in Alzheimer's disease model mice relaxes pericytes, improves cerebral blood flow and reduces immune cell stalling and hypoxia. Nat Neurosci. 2024 Nov; 27(11):2086-2100.
编 译 / 梁景兰

校 审 / 周立爽
声明:脑医汇旗下神外资讯、神介资讯、神内资讯、脑医咨询、Ai Brain 所发表内容之知识产权为脑医汇及主办方、原作者等相关权利人所有。
