



“重庆大学附属肿瘤医院神经外科月刊”每月回顾经典病例,针对神经系统疾病诊断、治疗提出最优治疗方案。病例包括神经系统肿瘤、脑血管疾病、脊柱脊髓疾病等。科室经历20余年发展,已形成以神经外科为先导、肿瘤专科为特色、专家有特长、专病专治的特点,拥有国内外先进技术并可以保持同步发展的神经外科专科队伍。科室目前可开展精准外科体系治疗脑胶质瘤、经鼻内镜下鞍区肿瘤切除手术、复杂颅内颅内动脉瘤介入及夹闭手术、脊柱脊髓肿瘤及退行性病变微创手术、神经导航或机器人辅助下脑内组织穿刺活检技术、神经内镜下脑室内、松果体区肿物切除、神经内镜与显微镜双镜联合行大脑深部、颅底的复杂肿瘤切除等。
本期为大家分享,重庆大学附属肿瘤医院神经外科病例精彩内容:多发脑膜瘤的分子机制及综合治疗。欢迎阅读和分享!
病例1
01
病史摘要
![]()
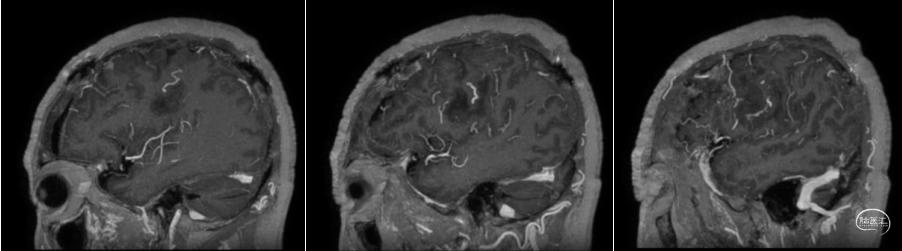
术前MRI
![]()
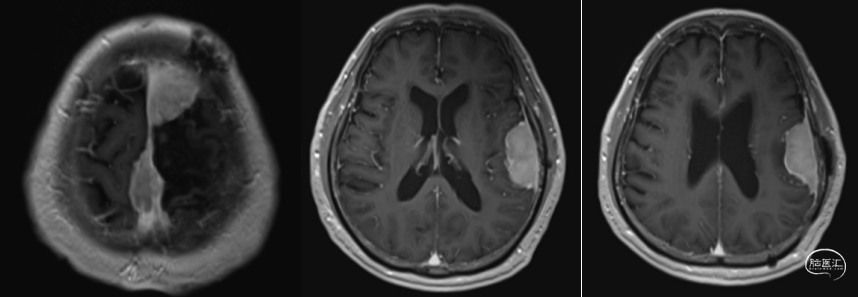
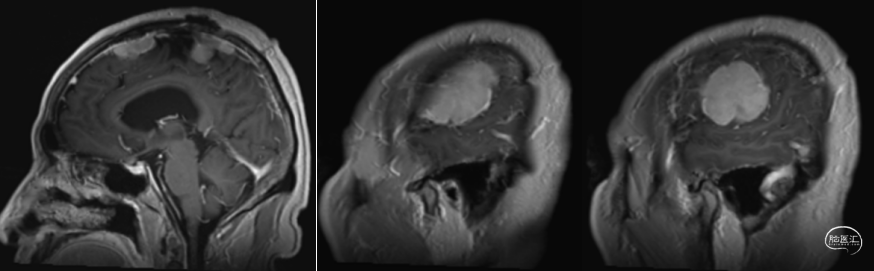
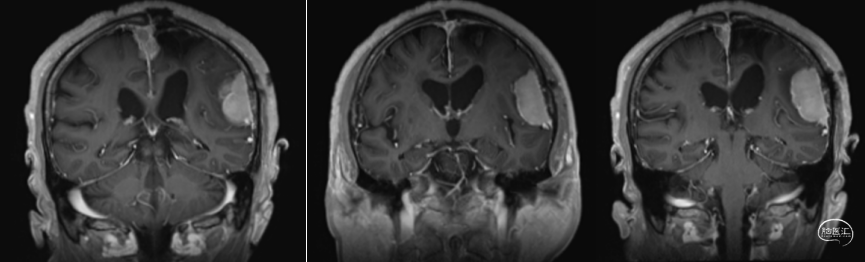
![]()
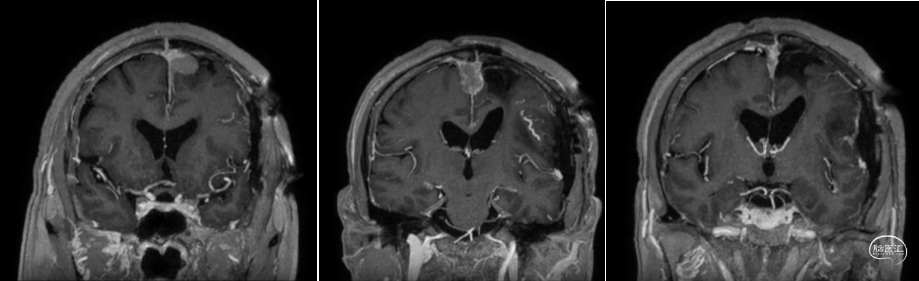
术后MRI
![]()
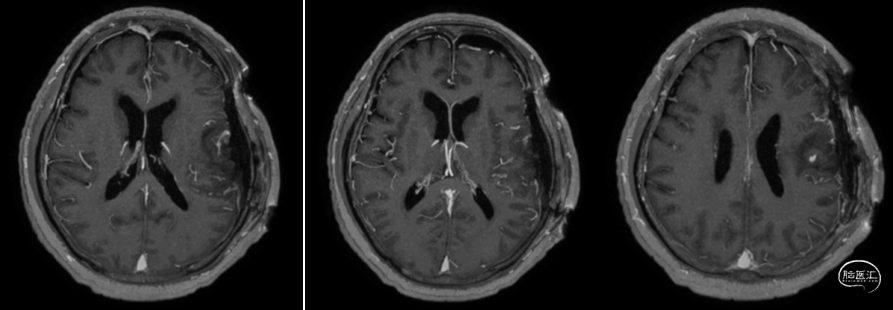
![]()
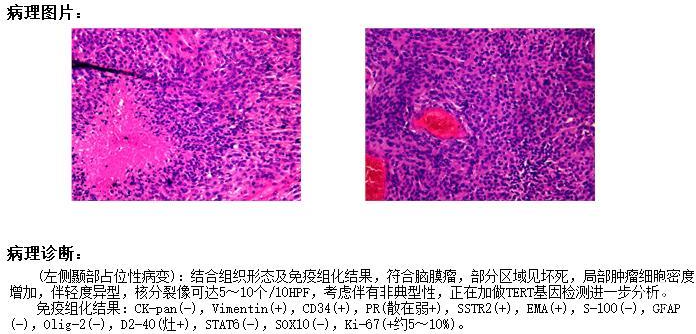
术后病理
![]()
病例2
02
![]()
病史摘要
![]()
![]()
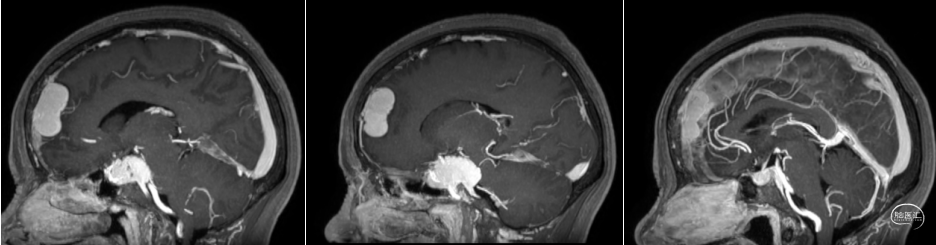
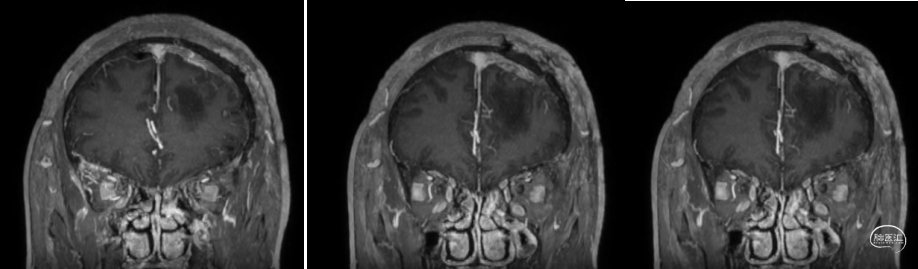
术前MRI
![]()
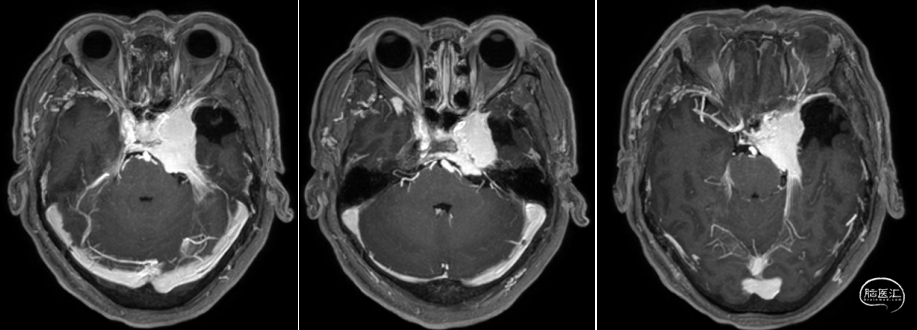
![]()
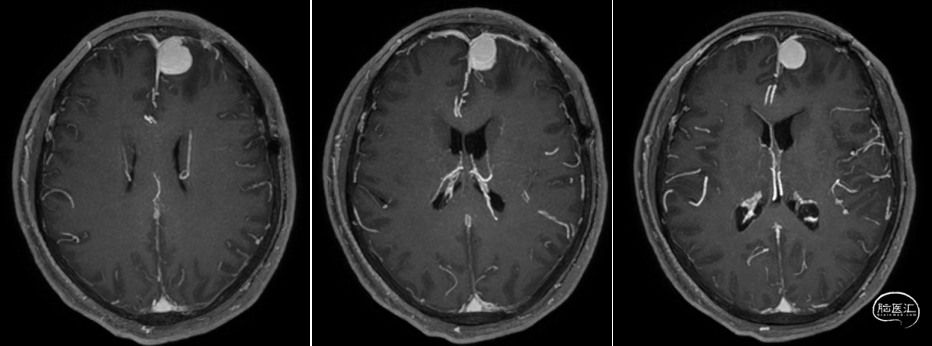
术后MRI
![]()

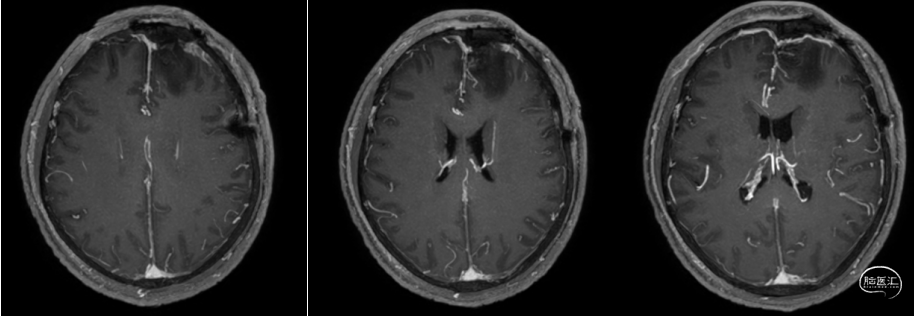
![]()
术后病理
![]()

讨论
脑膜瘤是中枢神经系统最常见的肿瘤,占总体的38.7%,其中绝大多数脑膜瘤是单发,多发脑膜瘤较为罕见,仅占总体的1%-3%[1]。多发脑膜瘤最早于1938年由Cushing和Eisenhardt提出,是指在没有神经纤维瘤和听神经瘤的情况下,颅内出现两个或两个以上空间上分隔的同时或异时性脑膜瘤。目前有关多发脑膜瘤的研究较少,大多是小样本研究和个案报道。近年来,多发脑膜瘤的发病率有升高趋势,其自然史、发病机制和治疗原则与单发脑膜瘤不完全相同,了解脑膜瘤多发的分子遗传学机制对其临床治疗和评估预后有重大意义,本次为大家简单介绍多发脑膜瘤的分子遗传机制的研究进展。
多发脑膜瘤的背景
多发脑膜瘤患者大多存在NF2病家族遗传史,散发的多发脑膜瘤患者常在童年时经历过电离辐射治疗或激素治疗。多发脑膜瘤位置多发于大脑凸面,数目在2-10个不等,体积较小,生长速度相对较慢[2],女性倾向高于单发脑膜瘤[3,4]。多发脑膜瘤组织学分型多良性,可手术切除,其预后较好,死亡率仅约3.4%,复发率与单发脑膜瘤无明显统计学差异[1,5]。多发脑膜瘤的发病机制尚不明确,其主要的起源假说有两种,一些研究表明多发脑膜瘤的肿瘤是各自独立散发的,因为在同一患者的不同脑膜瘤结节中,检测到的脑膜瘤组织学类型和细胞遗传学分析结果各不相同[6,7];另外一些研究认为,多发脑膜瘤是由单个脑膜瘤的细胞通过蛛网膜下腔的脑脊液传播到脑膜的不同位置形成的,因为大部分的多发脑膜瘤分布在大脑凸面[3,4],且同一患者的脑膜瘤多出现在同一侧大脑半球[8],另有文献报道的不同脑膜瘤结节的分子基因分析以及常规NF2基因突变检测结果也都支持该假说[9,10]。这两种假说可以解释不同的多发脑膜瘤案例,并都有相应的理论支持,多发脑膜瘤具体的发病机制还需要进一步的分子遗传学研究来证实。
目前WHO组织学分级依然是脑膜瘤诊断和治疗的金标准,然而近年来随着脑膜瘤的基因组学和表观遗传学的迅速发展,脑膜瘤致病的分子机制正逐步被阐述,目前已经证实单发脑膜瘤的发生与NF2、TRAF7、KLF4、AKT1、SMO和PORR2A等基因的缺失或突变相关[11,12],根据脑膜瘤的基因标志来分型更有利于其临床治疗,比如Sahm, et al. [13]发现将脑膜瘤分为6个DNA甲基化基团组,可以更精确地描述肿瘤的临床特征。但是由于多发脑膜瘤的分子致病机制尚不清楚,目前还是以WHO分型为金标准。在WHO组织学分级上,多发脑膜瘤的占比与单发脑膜瘤无明显统计学差异,都是以WHO I级为主,亚型以脑膜上皮型居多[5],其中同一多发脑膜瘤患者可有不同级别的脑膜瘤,而且同级别脑膜瘤中组织学亚型也可不同[7,14]。在美国2012-2016年报导的脑膜瘤案例中,WHO I级占80.5%,WHO II级占17.7%,WHO III级占1.7%[15];而在Tsermoulas, et al. [4]的研究中,多发脑膜瘤的WHO分型占比分别为WHO I级78.2%,WHO II级18.4%,WHO III级3.4%,其中20%的患者有不同级别的脑膜瘤。近年来有关多发脑膜瘤的研究较少,而其发病率却有逐年提升的趋势,阐述其分子致病机制成为刻不容缓的问题。
多发脑膜瘤的分子致病机制
NF2基因突变
NF2基因是经典抑癌基因,定位于22号染色体,包含17个外显子,其编码的merlin蛋白具有肿瘤抑制作用。NF2基因的功能丧失性(LOF)突变是多发脑膜瘤最常见的危险因素,占比可达83%。NF2基因突变的多发脑膜瘤可按其发病方式分为家族遗传病相关性和散发性。
II型神经纤维瘤病(NF2病)是常染色体显性遗传的家族遗传病,典型临床表现为双侧前庭神经鞘瘤。多发脑膜瘤常出现在NF2家族遗传病患者中,以NF2综合征的临床表现之一被发现。在芬兰的一个流行病学研究中,约20%的多发性脑膜瘤患者有NF2家族遗传病病史[16];但该结果可能低于实际值,因为该研究中NF2病的预计出生率为1/87410,而英国一个更大样本量的研究显示,NF2病的预计出生率为1/33000-40000[17]。在NF2病中,90%以上可以检测到胚系NF2基因突变,该突变也导致将近50%的NF2病患者有脑膜瘤表现[18]。NF2家族遗传病发生脑膜瘤的风险与NF2基因突变的类型和位置相关,截短性突变比非截短性突变发生的风险大,5’端基因突变比3’端基因突变发生的风险大[19];非NF2病相关的家族多发脑膜瘤病极为罕见,一般患者肿瘤和外周血中检测不到NF2基因突变,但其家属也需要测定基因图谱以评估患病风险[20]。
在散发性多发脑膜瘤中,也常会出现NF2基因突变[21],统计表明约2/3的散发性脑膜瘤存在NF2基因的体细胞双等位基因失活。在不同类型的散发脑膜瘤中,NF2基因突变发生的频率也不同,纤维母细胞性脑膜瘤和过渡型脑膜瘤中NF2基因突变频率分别为70%和83%,而上皮型脑膜瘤中NF2基因突变频率仅为25%[19],大多NF2病相关性多发脑膜瘤也表现为纤维母细胞型和过渡型。有文献报道,在NF2突变的脑膜瘤中常常会检测到18号染色体上的DAL-1(EPB41L3)基因缺失或突变,该突变使DAL-1基因表达下调,DAL-1蛋白功能缺失[22]。但是在小鼠实验中,DAL-1基因缺失并不会影响细胞的增殖和生长,也不会增加肿瘤患病风险[23]。DAL-1蛋白经常和merlin蛋白合并缺失,目前认为DAL-1基因突变可能是NF2基因突变的伴随效应。
SMARCB1基因突变
SWI/SNF染色质重塑复合体由9-12个亚基组成,包括一个ATP酶亚基(SMARCA2或SMARCA4)、进化保留核心亚基、SMARCB1、SMARCC1、SMARCC2和复合体特定变异亚基[24],它们协同启动、表达基因组上的基因。SMARCB1是神经鞘瘤病的易感基因,表现为发生在双侧前庭除外的颅内多发神经鞘瘤,该基因突变占家族性神经鞘瘤病的30-40%,但在散发性案例中占比不足10%。神经鞘瘤病可略微增加患脑膜瘤的风险,研究发现在神经鞘瘤患者中,约5%的人一生中会出现一个或多个脑膜瘤[25];而且NF2和SMARCB1都位于22号染色体上,相隔仅有6mb,在肿瘤发展过程中当体细胞22号染色体臂大部分或全部丢失时,这两个基因经常合并丢失[18],这也是怀疑SMARCB1是多发脑膜瘤的易感基因的原因。体细胞SMARCB1突变在散发性单发脑膜瘤中占比虽然不足3%,但是有位置偏好,肿瘤多发生在大脑镰部位[26,27]。在多发脑膜瘤中也存在体细胞SMARCB1突变,Wang, et al. [28]在一个外周血SMARCB1基因突变阴性的多发脑膜瘤患者中发现了SMARCB1的体细胞突变;Christiaans, et al. [29]在一个家族性多发脑膜瘤的家庭中发现了SMARCB1等位基因缺失的胚系突变,并提出了NF2和SMARCB1的四打击致病机制。然而有文献报道,在对45个非NF2相关性多发脑膜瘤患者筛查中并未发现种系SMARCB1基因突变。目前,SMARCB1基因在多发脑膜瘤发生发展中的作用还不明确,SMARCB1基因可能只是多发脑膜瘤一个偶然易感基因,其具体机制还有待进一步的研究证实。
SMARCE1基因突变
SMARCE1基因不存在于低等真核生物,仅存在于哺乳动物的SWI/SNF染色质重塑复合体中[30]。最近SMARCE1基因突变被提出可能与多发脑膜瘤相关,但是该突变似乎特定引起透明细胞亚型脑膜瘤,而且几乎所有被发现的SMARCE1突变都是截短突变,并在外显子5-9之间发生,表明该区域可能与SMARCE1蛋白的表达和其完整性密切相关[18]。有研究在三个患有多发脊髓透明细胞脑膜瘤的家族中发现了SMARCE1染色质重塑因子的种系功能缺失突变[31],随后种系SNARCE1突变在颅内多发脑膜瘤中被发现[32];体细胞SMARCE1基因丢失也在一个被临床诊断为NF2病多发脑膜瘤患者的肿瘤细胞中检测到。SMARCB1和SMARCE1都有DNA结合特性,但两者对SWI/SNF复合物的DNA结合特性都不是必需的,它们在基因调控中的作用及其参与脑膜瘤发展的机制还需要进一步研究。
其他基因突变
在一个芬兰的遗传性多发脑膜瘤大家族中,检测到了SUFU错义突变(c.367C>T)[33],但是该突变在一个对121个多发脑膜瘤患者的扩大性基因筛查中并没有被检测到。CCM3基因突变也可能与多发脑膜瘤的发生相关[34],而且在小鼠实验中切除CCM3基因可以使AKT信号通路失活,增强小鼠细胞的增殖和抗凋亡能力[35]。近期还有研究发现,一些肿瘤综合征(如Werner综合征[36]、Cowden综合征[37]等)能增加多发脑膜瘤的患病概率,但是这些肿瘤综合征的患者有些接受过激素治疗,目前激素和肿瘤综合征在这些患者的致病因素中所占的比重尚不清晰。
结论
多发脑膜瘤与单发脑膜瘤的分子致病机制不尽相同,目前在单发脑膜瘤中,测序技术已经逐步应用到临床医疗诊断中,通过检测并分析患者脑膜瘤标本中特定的基因的点突变、插入、缺失、融合、拷贝数变异等情况,可帮助病人选择获益最佳的治疗方案。而由于多发脑膜瘤的发病率较低,目前我们的多发脑膜瘤的分子遗传学特征了解还不够全面,进一步地研究其分子致病机制有助于我们对多发脑膜瘤的临床诊断和预后评估。
治疗建议
多发脑膜瘤患者的治疗策略必须定制,因为肿瘤的性质和治疗对不同患者的潜在后果差异很大。多发脑膜瘤患者的总体目标是肿瘤控制,手术切除全部肿瘤是不可能的,需要有慢性病的视角。这种多样性带来了特殊的挑战,包括定位症状病变,选择最合适的治疗方式,避免治疗风险,以及预测个体未经治疗的肿瘤的行为。至关重要的是要考虑到MM患者可能需要在其一生中反复治疗。
重要的考虑因素是任何肿瘤的可能生长速度。Wong等2013对MM的自然病史和生长速率进行了研究。他们分析了12例患者的55个肿瘤,平均随访61个月(24-101个月)。他们报告的平均生长率为0.46cm3/年(范围0.57-2.94cm3/年),这与意外偶然发现的单发脑膜瘤(SM)的报告相似。对肿瘤多样性与生长率之间的关系也进行了分析,但未观察到每位患者脑膜瘤的数量与生长率之间的相关性。
虽然脑膜瘤很常见,但其治疗的证据水平却低得惊人。管理似乎是标准化的,但对照临床试验并不常见,因此医疗标准在很大程度上是由当地经验、长期传统和偶尔基于经验的实践来定义的。此外,在许多情况下,一种以上的治疗方案是合理的。2016年,欧洲神经肿瘤协会(EANO)发布了首份关于脑膜瘤诊断和治疗的指南。自那时以来,用于诊断和临床决策的证据水平有所提高,包括来自对照临床试验和WHO中枢神经系统肿瘤分类新第5版的数据。因此,EANO在2021年更新了其脑膜瘤治疗指南。
观察
越来越多的意外偶发、无症状脑膜瘤通过神经影像学诊断,需要有针对性的治疗和随访策略的发展。大多数脑膜瘤属于WHO 1级,由于与治疗相关的并发症发生率不可忽视,因此很难在这一阶段推荐治疗。在大多数患者中,临床或影像学进展发生在诊断后的5年内,islam等(2019)认为,在此之后可能不太需要定期监测。
基于这些发现,我们开发了一种预后模型来指导意外偶发性脑膜瘤患者的个性化监测,其中包括单发脑膜瘤和多发脑膜瘤。该模型结合了患者年龄、表现状态、合并症和MRI特征的数据,将患者分为低、中、高风险的生长和进展,从而制定个性化的监测策略。肿瘤高信号、体积增大、靠近关键神经血管结构、肿瘤周围信号改变等因素均增加了诊断后5年内疾病进展的风险。然后可以根据预测的疾病进展率对患者进行临床和影像学随访。此外,研究表明,大多数意外偶发脑膜瘤在随访期间保持稳定,并在5年后达到生长平台期。最后,该模型对有合并症的低风险和老年患者的严格监测几乎没有好处,因为他们在估计寿命内不太可能需要干预,因此不建议继续进行影像学监测。
因此,越来越多的证据表明,对很大一部分患有单发脑膜瘤和多发脑膜瘤的患者不需要积极治疗。对于绝大多数患者,这导致行定期监测,包括重复的磁共振扫描。多发脑膜瘤采用这种策略的证据并不像单发脑膜瘤那样有力,但足以表明,无症状多发脑膜瘤患者可以通过连续影像学检查进行安全治疗,直到影像学或症状性生长。与EANO一致,对于疑似或WHO 1级脑膜瘤,建议每年进行MRI扫描5年。此后,间隔可以翻倍。根据Islam等(2020)的研究结果,个性化随访似乎是合理的,特别是对于有合并症的老年患者。
手术
手术是大多数单发脑膜瘤和多发脑膜瘤患者的症状性或生长性脑膜瘤的主要治疗方式,旨在缓解肿块占位效应和神经系统症状,并获得组织诊断。尽管缺乏将手术与其他疗法进行比较的随机试验,但这是一种行之有效的方法。手术作为一线治疗的证据源于许多病例系列,表明切除程度(EOR)是一个重要的预后因素。脑膜瘤手术的主要目标是最大限度的安全切除,低并发症发生率和保留神经和认知功能。如果可能的话,目标是大体全切除(GTR),但EOR还取决于许多因素,包括肿瘤的位置、一致性和大小,以及靠近或累及关键神经血管结构。对多发脑膜瘤患者的原则相同,但手术决策更具挑战性,因为切除一个以上的肿瘤可能是必要的,尽管切除所有肿瘤很少是一个合理的目标。手术成功后,肿瘤可能会留下,因为手术应该集中在有症状的或正在生长的肿瘤上。因此,多发脑膜瘤患者很少能像单发脑膜瘤患者那样通过手术“治愈”。当GTR不是一个现实的目标时,目标应该是安全的次全切除并保留神经和认知功能。残余肿瘤的处理应个体化,包括监测、术后适形分割放疗或立体定向放射外科(SRS)治疗。
在手术前,与患者讨论手术目的以及手术风险是至关重要的。对于多发性脑膜髓瘤患者,明确解释治疗目标是很重要的,特别是在疾病控制是目的,并非所有肿瘤都可以切除的情况下。患者应做好接受多种治疗和终身监测的准备。
放射外科治疗
立体定向放射治疗对于小肿瘤、高手术并发症发生率部位的肿瘤、老年人或身体不适的患者是手术治疗的重要替代方法。Bir等(2017)研究表明,SRS对直径小于或等于3cm的颅内小脑膜瘤的局部控制与Simpson 1级切除术相当。他们还表明,与不完全切除的患者相比,SRS组的无复发生存期较长。因此,小脑膜瘤不适合次全切除术。然而,术后症状改善的可能性较大。此外,2项回顾性研究显示,SRS或大分割放射外科治疗后肿瘤大小的减小预示着5年和10年后肿瘤的长期控制。在这些系列中,10年无复发生存率分别为93.4%和95.7%。
由于同时治疗空间分离的肿瘤并获得良好的长期效果是可能的,SRS在MM患者中发挥了更重要的作用。特别是对于颅底脑膜瘤累及颅神经和血管,GTR很少可行,越来越多地采用次全切除术和SRS治疗的多模式入路。因此,联合治疗方式对脑膜瘤的作用将会增加,特别是对多发脑膜瘤患者。SRS治疗后需要考虑的重要并发症包括瘤周脑水肿、放射诱导的神经病变和内分泌病变。
分割外放射治疗
分割外放射治疗继续有助于脑膜瘤的治疗。对于不能安全切除或不能完全切除的脑膜瘤患者,这是一种公认的治疗方法。多项试验表明,对毒性可接受的WHO2级和3级脑膜瘤患者进行分割放疗的益处。在一项对WHO 2级和3级脑膜瘤进行手术切除和/或放疗的回顾性队列研究中,WHO 2级脑膜瘤患者的5年总生存率为75.9%,WHO 3级脑膜瘤患者的5年总生存率为55.4%。此外,在WHO 2级脑膜瘤患者中,大体全切除和术后放疗是提高生存率的独立预测因素。
既往接受过放射治疗的脑膜瘤患者是一个具有挑战性的队列,因为先前暴露的组织的再照射耐受性较低。现有的数据非常有限,但一些论文表明,在治疗复发性脑膜瘤的特定患者中,可以根据以前的剂量分布、初次照射和再程照射之间的时间以及位置,特别是在危及器官附近,进行再程照射。很明显,分割放疗可以避免额外的外科手术,但有长期毒性的风险,包括神经认知障碍、垂体功能减退和辐射诱发的肿瘤。然而,在过去的几十年里,成像、靶区勾画和三维规划方面的技术进步改善了肿瘤覆盖范围和规避关键结构,使放射更加准确、有效和安全。
药物治疗
目前脑膜瘤还没有有效的药物治疗方法,关于治疗前景的讨论超出了本文的讨论范围。如果可行,MM患者将特别受益,因为可以一次性治疗所有肿瘤。在缺乏积极的临床试验和进一步研究的情况下,很难提出建议或预测。
认知功能
多项研究表明脑膜瘤患者在术前和术后均存在认知功能障碍。记忆力、注意力和执行功能最常受到影响。术前损伤可能是由于解剖位置、社会心理因素、癫痫及其治疗或肿瘤或与肿瘤相关水肿引起的颅内压升高所致。复杂性在多发脑膜瘤患者中增加,因为可能不清楚哪个肿瘤或哪些肿瘤导致认知功能障碍。即使没有因果关系,认知障碍与额叶或颞叶肿瘤位置、肿瘤大小和脑水肿体积之间也存在相关性。手术后认知功能正常改善。癫痫发作和抗癫痫药物可能是术后认知障碍的原因。令人惊讶的是,很难证明肿瘤偏侧性与术后认知功能之间有任何明确的相关性。术后长期认知障碍见于术前脑水肿的脑膜瘤患者。有趣的是,即使经过长时间的随访,也没有发现放射治疗与认知功能之间的相关性。在多发性脑膜瘤患者中,多种治疗、残留肿瘤、癫痫治疗和社会心理压力因素的影响可能是相关的,并且可能是累积的。
生活质量
近年来,人们越来越多地认识到脑膜瘤患者的HRQoL降低。脑膜瘤患者术前HRQoL受损,即使手术后神经功能缺损得到改善,长期HRQoL的降低也可能发生在认知、情感和社会领域,并伴有睡眠障碍、疼痛、焦虑和疲劳。此外,能够开车或重返工作岗位的患者数量随着时间的推移而减少,这是一个重大的社会经济负担。较差的HRQoL与肿瘤大小大、WHO分级高、肿瘤复发、诊断后时间短、年龄≥50岁、创伤后应激、人格改变、精神错乱、左半球肿瘤位置、头痛和癫痫发作有关。不言而喻,在MM患者中,对于认知功能障碍,与SM患者相比,多发性肿瘤的影响及其治疗可能对HRQoL产生更深远的影响,但缺乏相关研究。
预后
据我们所知,2020年的SEER研究是首次对多发脑膜瘤患者进行大队列生存分析,并显示病变数目、诊断年龄和性别影响多发脑膜瘤患者的OS。单发脑膜瘤患者的中位生存期为180个月,多发脑膜瘤患者的中位生存期为94个月。分析显示,每增加一个病变,OS就会逐渐减少。与未接受放射治疗的患者相比,接受过放射治疗的患者有较长的生存期。女性患者的OS较长。对男性队列的分析显示,多发脑膜瘤的生存期从41岁开始降低,每增加10年生存期缩短。对女性队列的类似分析表明,多发脑膜瘤从51岁开始减少OS,每增加10岁,OS就会缩短。由于这些结果仅代表一项研究,因此适用性有限,应谨慎应用。
结论
已发表的关于多发脑膜瘤的资料很少,大多局限于病例报告和小病例系列。尽管如此,我们回顾了多发脑膜瘤文献,并对该主题进行了最广泛和全面的回顾,包括基于证据的管理范式,这将作为临床决策和未来研究的有力基线。目前的流行病学数据表明,多发脑膜瘤的真实发病率比以前认为的要高得多,这表明需要进一步的研究和更成熟的治疗建议。即使单发脑膜瘤和多发脑膜瘤之间有一些明显的相似之处,如年龄和性别分布、肿瘤分级和生长速度,但也有明显的差异,如潜在遗传疾病的可能性、治疗目标和预后。多发脑膜瘤患者的管理是复杂的,包括多种治疗,有时采用不同的方式,并终身监测。我们提倡将多发脑膜瘤作为一种慢性病来对待和管理。
参考文献
专家简介

杨海峰 主任医师
重庆大学附属肿瘤医院神经外科科主任,主任医师,医学博士,硕士研究生导师
硕士毕业于首都医科大学北京天坛医院
博士毕业于首都医科大学宣武医院
主要从事神经系统肿瘤的显微手术及内镜手术治疗
中国抗癌协会肿瘤神经病学专委会常委
中国解剖学会神经解剖专委会委员
中国临床肿瘤学会神经肿瘤专委会委员
中国医学装备协会神经外科分会委员
中国医药创新促进会脑神经药物临床研究专业委员会委员
中国脑膜瘤多学科诊疗协作组专家委员会委员
重庆市医师协会脑胶质瘤专委会副主任委员
重庆市医药生物技术协会神经外科专委会主任委员
重庆抗癌协会神经肿瘤专委会副主任委员
中国临床医生杂志审稿专家
Cleveland Clinic访问学者
近年来承担及参与国家级、省部级5项;发表国际SCI高质量论文及国内核心期刊20余篇。主要研究脑肿瘤精准手术治疗体系及脑肿瘤发生发展机制研究

点击/识别二维码,前往杨海峰 主任医师学术主页
查看更多精彩内容

王世强 副主任医师
医学博士,重庆市肿瘤医院神经外科副主任医师
重庆市医学会神经内镜专业委员会委员曾先后在中国武警脑科医院、上海交通大学第一人民医院进行系统的显微及内镜解剖培训,曾赴意大利米兰Neurologico Carlo Besta神经医学中心参观学习
参加工作以来以第一作者发表SCI文章3篇,获得发明专利1项,主持及参与市局级课题3项
擅长颅脑肿瘤(脑膜瘤、胶质瘤、垂体瘤、颅咽管瘤、转移瘤、听神经瘤、生殖细胞肿瘤等)、椎管内肿瘤、脑积水和脑血管疾病的显微外科手术及综合治疗

点击/识别二维码,前往王世强 副主任医师学术主页
查看更多精彩内容
科室简介

01
科室简介:
02
科室人员构成:
03
科室特色:
(1)现代精准外科体系治疗脑胶质瘤;
(2)经鼻内镜下鞍区肿瘤切除手术,包括常见垂体腺瘤、颅咽管瘤、鞍结节脑膜瘤、侧颅底肿瘤等;
(3)复杂颅内颅内动脉瘤介入及夹闭手术;
(4)神经导航或机器人辅助下脑内组织穿刺活检技术;
(5)神经内镜下脑室内、松果体区肿物切除以及神经内镜下三脑室底造瘘技术治疗脑积水;
(6)脊柱脊髓微创切除及固定手术;
(7)神经内镜与显微镜双镜联合行大脑深部、颅底的复杂肿瘤切除。

点击/扫描上方二维码
查看重庆大学附属肿瘤医院神经外科更多内容
![]()
声明:脑医汇旗下神外资讯、神介资讯、神内资讯、脑医咨询、Ai Brain 所发表内容之知识产权为脑医汇及主办方、原作者等相关权利人所有。
投稿邮箱:NAOYIHUI@163.com
未经许可,禁止进行转载、摘编、复制、裁切、录制等。经许可授权使用,亦须注明来源。欢迎转发、分享。
