


本文来源于公众号:小崔和他的小伙伴们

结果:1.AD患者大脑中髓鞘丢失
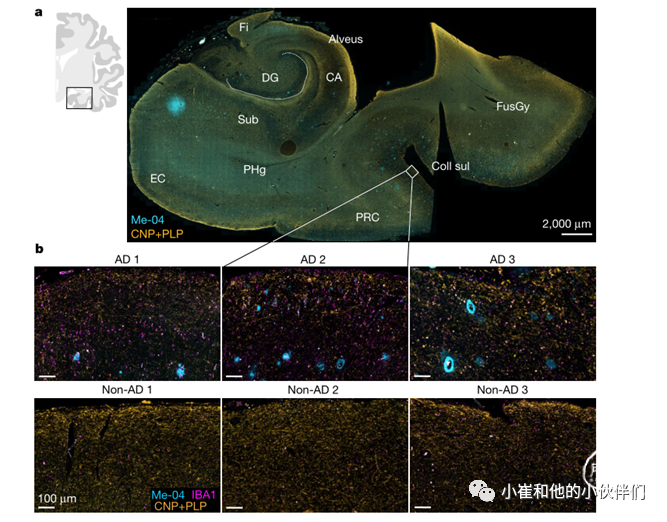 图1:AD患者的髓鞘损伤
图1:AD患者的髓鞘损伤结果2:髓鞘缺陷加剧 Aβ 沉积
为了确定这种与衰老相关的髓鞘缺陷是否会导致淀粉样蛋白沉积,作者将 Cnp−/− 和 Plp−/y 突变体与 5×FAD 小鼠杂交,并分析了所得后代的斑块负担。作者团队基于光片显微镜 (LSM) 的 iDISCO 技术,优化了刚果红斑块染色和清除方案,以公正的方式确定整个大脑中的淀粉样蛋白斑块含量。结果发现与 5×FAD 小鼠相比,Cnp−/−;5×FAD 小鼠和 Plp−/y;5×FAD 小鼠在 6 个月龄时海马白质(海马槽)和皮质中的淀粉样斑块负荷显着增加(图 2a-d)。在这两种小鼠模型中,海马槽(海马白质束)的影响最强:在这里 Aβ 沉积在非常小的聚集体中,表明淀粉样蛋白种子的形成增加。当Cnp−/−小鼠和Plp−/y小鼠仅表现出轻微的髓鞘缺陷和反应性胶质增生时,在 6 个月大时观察到斑块负荷增加,但在 3 个月大时尚未观察到。此外,5×FAD对照小鼠和髓鞘突变小鼠在这个年龄的海马槽中没有表现出病理斑块。为了排除这些效应是 5×FAD 模型和 App 过度表达所特有可能性,作者在 Cnp−/− 小鼠和APPNLGF敲入模型的杂交小鼠中进行了验证。作者还测试了老年髓鞘突变动物本身(没有人类APP转基因)是否会在其有生之年自然形成淀粉样斑块。在14个月大的PLP−/y小鼠或22个月大的长寿小鼠(Emxcre Plp fl/fl小鼠)中都没有检测到淀粉样蛋白沉积。这些结果与啮齿动物淀粉样蛋白相对不容易聚集的研究结果相一致,表明过度表达啮齿动物Aβ的小鼠终生不会发生淀粉样变性。
继发性髓鞘改变已经在AD的小鼠模型中已有描述。然而,作者团队使用免疫染色、免疫印迹和电子显微镜(EM)分析没有在6个月大的5×FAD小鼠或APPNLGF小鼠中检测到实质性的髓鞘病理。同样,在CNP−/−小鼠和PLP−/y小鼠中发生的微小的髓鞘病理并没有通过杂交到5×FAD基因型而改变。因此,观察到的促进斑块的作用不仅仅是少突胶质细胞或髓鞘对淀粉样蛋白沉积的改变的二次反应。

结果3:急性脱髓鞘导致Aβ沉积
为了证实髓鞘功能障碍是斑块病理的上游驱动因素,作者测试了急性脱髓鞘的效果。幼年5×FAD小鼠喂饲含cuprizone(一种铜螯合剂)饲料4周,恢复期4周后,用激光共聚焦显微镜测定斑块载量。结果发现这种方法会导致内侧胼胝体和海马槽出现大量脱髓鞘,并伴有小胶质细胞增多。然而,解释这项实验的结果是复杂的,因为络合铜的性质干扰了斑块核心的形成(斑块核心的形成依赖于铜)。事实上,LSM的整体结果显示,cuprizone治疗似乎改善了海马和皮质Aβ病理学。然而,在海马槽等脱髓鞘严重的区域,淀粉样变性导致的脱髓鞘效应在螯合铜的抑制作用中占主导地位。在该大脑区域,用Aβ抗体对cuprizone处理的5×FAD小鼠的Aβ斑块进行染色发现淀粉样小聚集体的大量增加(图2e)。
5×FAD小鼠诱发实验性自身免疫性脑脊髓炎(EAE)的结果提供了进一步的独立证据。作者用髓鞘少突胶质细胞糖蛋白(MOG)肽对幼年5×FAD小鼠进行免疫,并分析4周后的大脑和脊髓(图2f)。先前报道对老年J20和Tg2576 AD小鼠模型的研究结果显示,EAE降低了脑斑块负荷。然而,由于大脑中缺乏淀粉样蛋白沉积病理,作者没有发现5×FAD EAE小鼠中淀粉蛋白沉积的差异。与此相反,在脊髓中脱髓鞘性EAE病理显著发展,并在病变周围环境中出现了小的非典型淀粉样蛋白聚集体,而对照5×FAD小鼠中没有这种聚集体(图2f)。作者通过Me-04对脊髓切片进行染色验证了聚集淀粉样蛋白的存在(图2f)。值得注意的是,在患有EAE的WT动物的脊髓中,没有发现这样的Me-04+物质,排除了在脱髓鞘病变中非特异性检测到脂质沉积的可能性(图2f)。综上所述,髓鞘缺陷(包括慢性和急性)驱动了AD小鼠模型中淀粉样蛋白沉积,表明髓鞘功能障碍是淀粉样蛋白沉积的上游风险因素。
结果4:髓鞘缺乏可改变 Aβ 沉积
结果5:髓鞘功能障碍影响APP代谢
接下来作者研究了髓鞘缺陷是如何驱动淀粉样变性的。理论上,这些缺陷可以促进APP加工和Aβ的产生,也可以干扰Aβ的去除(或两者兼而有之)。作者首先研究了Cnp−/−;5×FAD小鼠的APP代谢。APP染色呈阳性的轴突肿胀是缺血、脑损伤和髓鞘疾病的显著特征,作者推测这些肿胀有助于髓鞘突变动物体内产生Aβ。轴突运输的停滞可能通过增加轴突运输囊泡中BACE1和APP的相互作用而增加淀粉样蛋白的产生,轴突似乎是淀粉样蛋白分泌的重要部位。在Cnp- / -小鼠和Cnp- / -;5×FAD杂交小鼠中都观察到许多轴突起源的髓鞘损伤相关肿胀。在5×FAD对照小鼠中几乎没有发现这种肿胀,轴突损伤仅限于斑块附近并以环状模式排列(称为冠状) 。高压冷冻电镜分析显示,髓鞘损伤相关的肿胀富含囊泡结构,可能是内质体或溶酶体(图3a),这被认为是Aβ的主要产生位点。使用针对APP加工酶的抗体和多种APP-和Aβ-特异性抗体(图3b),作者发现Cnp−/−;5×FAD小鼠大脑中的轴突肿胀处积累了β-分泌酶和γ-分泌酶,APP和BACE1共染色,因此β-和γ-裂解的APP片段和Aβ染色阳性(图3c–e)。免疫染色还显示 Cnp−/−;5×FAD 小鼠皮质组织中可溶性 APPβ 水平增加。在这个大脑区域除了斑块相关的肿胀之外,还观察到许多与白质类似的髓鞘损伤相关的肿胀。

结果6: 髓鞘缺陷改变小胶质细胞反应
神经胶质细胞在髓鞘碎片和淀粉样肽的清除中发挥重要作用,小胶质细胞在淀粉样斑块周围形成屏障。因此,作者在体外和体内研究了这些细胞在受到髓鞘缺陷挑战时如何对淀粉样蛋白做出反应。预先将骨髓来源的巨噬细胞(一种研究AD相关吞噬细胞功能的常用体外模型)暴露在髓鞘碎片中,会强烈地抑制了淀粉样蛋白的吞噬作用。在体内,尽管Cnp−/−;5×FAD 小鼠和 Plp−/y;5×FAD 小鼠中小胶质细胞未能聚集在淀粉样斑块周围,但白质和灰质的小胶质细胞数量都大幅增加(图 4a-d)。

为了更好地了解髓鞘缺陷诱导的DAM样小胶质细胞特征,作者进一步对Cnp−/-;5×FAD小鼠和各自对照的脑组织进行了单核RNA-seq(snRNA-seq)(图5a)。聚类分析确定了五个主要的小胶质细胞和巨噬细胞亚群,包括两个不同的具有DAM标记基因高表达的簇(簇4和簇5)(Trem2、Lpl和Spp1)(图5b–d)。簇4几乎完全来源于具有5×FAD基因型的细胞,而簇5由Cnp−/−背景的细胞组成(图5e)。因此,作者将这两个簇分别称为髓鞘DAM和淀粉样蛋白DAM。髓鞘DAM和淀粉样蛋白DAM之间的差异表达分析显示,在髓鞘DAM中脂质代谢相关基因(Apoe、Abca1和Apobec1)和Ms4a簇的基因上调,这与批量RNA-seq实验中的发现相吻合(图5f)。淀粉样蛋白DAM中特异性上调的基因包括Cst3、Ctna3(编码α-T-连环蛋白)和Gpc5(编码硫酸乙酰肝素蛋白聚糖5)。尽管检出率较低,但Cst7也特异性富集于淀粉样蛋白DAM中(图5g)。值得注意的是,当比较CNP−/−;5×FAD小鼠和5×FAD小鼠在淀粉样蛋白DAM簇内的小胶质细胞时,作者仍然观察到CNP−/−;5×FAD小胶质细胞的APOE上调。对 DAM 簇之间共有的DEG和DAM簇内基因型比较的分析表明,Cnp−/−;5×FAD杂交小鼠的淀粉样蛋白DAM和髓鞘DAM均保留了另一个DAM簇的特征,这突出了该小鼠模型中小胶质细胞的中间表型。

结果7:髓鞘老化是 AD 的危险因素
Depp C, Sun T, Sasmita AO, Spieth L, Berghoff SA, Nazarenko T, Overhoff K, Steixner-Kumar AA, Subramanian S, Arinrad S, Ruhwedel T, Möbius W, Göbbels S, Saher G, Werner HB, Damkou A, Zampar S, Wirths O, Thalmann M, Simons M, Saito T, Saido T, Krueger-Burg D, Kawaguchi R, Willem M, Haass C, Geschwind D, Ehrenreich H, Stassart R, Nave KA. Myelin dysfunction drives amyloid-β deposition in models of Alzheimer's disease. Nature. 2023 Jun. 618(7964):349-357.
![]()
声明:脑医汇旗下神外资讯、神介资讯、神内资讯、脑医咨询、Ai Brain 所发表内容之知识产权为脑医汇及主办方、原作者等相关权利人所有。

