加拿大多伦多大学附属森尼布鲁克健康科学中心神经病学科的Mary Jane Lim-Fat等根据文献和专家意见,提出AYA人群CNS肿瘤分子检测的共识指南,发表在2022年9月的《Frontiers in Oncology》在线。
——摘自文章章节
【Ref: Lim-Fat MJ, et al. Front Oncol. 2022 Sep 23;12:960509. doi: 10.3389/fonc.2022.960509. eCollection 2022.】
2016年世界卫生组织(World Health Organization,WHO)中枢神经系统肿瘤分类修订版引入分子遗传学特征以补充组织学诊断和分级;经过中枢神经系统肿瘤分类分子信息及实践方法联盟(cIMPACT-NOW)系列更新,2021年WHO中枢神经系统肿瘤分类第五版(WHO CNS5)将分子诊断与组织形态和免疫组织化学等建立的肿瘤诊断方法相结合。美国国家癌症研究所将15-39岁的人群定义为青少年和青年(adolescent and young adult,AYA)。AYA中枢神经系统肿瘤有许多不同的亚型,包括胶质瘤(成人和儿童类型)、髓母细胞瘤/胚胎性肿瘤和室管膜瘤。目前,对于这个年龄段的患者诊断或治疗没有明确的指南。加拿大多伦多大学附属森尼布鲁克健康科学中心神经病学科的Mary Jane Lim-Fat等根据文献和专家意见,提出AYA人群CNS肿瘤分子检测的共识指南,发表在2022年9月的《Frontiers in Oncology》在线。
WHO CNS5将胶质瘤、胶质神经元肿瘤、神经元肿瘤分为6个不同亚型:成人型弥漫性胶质瘤、儿童型弥漫性低级别胶质瘤、儿童型弥漫性高级别胶质瘤、局限性星形细胞胶质瘤、胶质神经元和神经元肿瘤以及室管膜瘤。对成人型弥漫胶质瘤和儿童型弥漫胶质瘤的临床和分子区别是WHO CNS5中最重要的变化。成人和儿童胶质瘤分层见表1。
表1. 推荐AYA胶质瘤患者检测生物标志物及其临床意义。
在WHO CNS5中,对成人型弥漫性胶质瘤进行肿瘤类型内分级而不是跨肿瘤类型分级。从之前的15种类型,划分成现在的3种:星形细胞瘤,异柠檬酸脱氢酶(isocitrate dehydrogenase,IDH)突变型、少突胶质细胞瘤;IDH突变伴1p/19q联合缺失型、胶质母细胞瘤;IDH野生型。在IDH突变型胶质瘤中,ATRX缺失足以诊断星形细胞系肿瘤,而无需进行1p/19q共缺失分析。相反,对ATRX基因表达的IDH突变型胶质瘤患者都应进行1p/19q共缺失分析。
所有IDH突变型弥漫性星形细胞瘤列为单一类型-星形细胞瘤,IDH突变可分为2、3、4级(不再使用间变性一词)。即使没有微血管增生或局灶坏死,细胞周期蛋白依赖性激酶抑制剂(cyclin-dependent kinase inhibitor,CDKN)2A/B纯合子缺失预示预后较差,被归类为星形细胞瘤,IDH突变,CNS WHO 4级。在WHO CNS5中,胶质母细胞瘤(glioblastoma,GBM)不再代指IDH突变型星形细胞胶质瘤。具有野生型IDH和组蛋白H3状态并伴有坏死或微血管增生的星形细胞瘤归类为IDH野生型WHO 4级胶质母细胞瘤。无坏死或微血管增生时,应评估EGFR扩增、端粒酶逆转录酶(TERT)启动子突变和/或7号染色体扩增和10号染色体缺失(Chr7+/10-)等遗传学改变,若存在其中一个以上改变,则肿瘤应归类为IDH野生型胶质母细胞瘤。值得注意的是部分弥漫型星形细胞瘤(WHO 2级)伴有单独TERT启动子突变。大多数GBM,特别是具有典型组织学特征的GBM,在老年人多见。在没有以上三种基因改变的IDH野生型弥漫型星形细胞瘤中,特别是在AYA患者中,需考虑儿童型胶质瘤。胶质肉瘤和巨细胞胶质母细胞瘤等亚型已不再列明。
儿童型弥漫性低级别胶质瘤(low-grade gliomas,LGG)WHO CNS5中儿童型弥漫性低级别胶质瘤被细分为弥漫性星形细胞瘤,伴MYB或MYBL1改变、丝裂原活化蛋白激酶(MAPK)通路改变的弥漫性胶质瘤(通常是BRAF或FGFR1改变)、血管中心型胶质瘤(MYB-QKI融合)、青少年多型性低级别神经上皮肿瘤(FGFR2融合)。从组织病理学角度来看,伴MAPK通路改变的儿童型低级别胶质瘤(low-grade gliomas,LGG)属于弥漫性胶质瘤,细胞密度低,而且具有轻度异型性。通常表现为OLIG2免疫阳性,GFAP不同程度表达。伴MYB或MYBL1改变的肿瘤由相对单一的胶质细胞起源的细胞组成,在纤维基质中有微圆形或纺锤状核,可能具有模糊的血管中心极性。常表现为GFAP阳性,OLIG2阴性。
儿童型弥漫性高级别胶质瘤(high-grade gliomas,HGG)与成人型胶质瘤(可能由低级别肿瘤转化而来)相反,儿童型HGGs由不同分子驱动产生。在AYA组内,儿童型弥漫性HGGs可进一步细分为弥漫性中线胶质瘤(diffuse midline gliomas,DMGs),伴H3 k27突变、弥漫性半球形胶质瘤,伴H3 G34突变、弥漫性儿童型HGG,H3及IDH野生型。儿童和青少年弥漫性中线胶质瘤(脑干、丘脑或脊髓)主要是由于编码H3.3或H3.1组蛋白的基因(分别为H3-3A、H3C2)出现杂合突变,导致第27位赖氨酸被蛋氨酸取代。显微镜下,DMGs呈星形细胞状,但也可表现出各种细胞学特征,如毛状、少突胶质、巨细胞、上皮样或未分化,有丝分裂指数升高,微血管增生和/或坏死,但不影响DMGs患者预后。在免疫表型上,DMGs可表达OLIG2、MAP2、S100以及不同程度的GFAP表达。
H3-G34突变型弥漫性半球胶质瘤通常发生在年龄较大的儿童和青少年中,有研究报道中位年龄为25岁。具有胶质母细胞瘤的组织学特征,典型的组织学形态为高级别的浸润性星形细胞瘤,核分裂常见,伴微血管增生和(或)坏死。部分肿瘤细胞形态类似中枢神经系统胚胎性肿瘤,或可见排列一致、密集的小蓝圆细胞,无明显微血管增生或坏死。其分子病理学特征为GFAP阳性、ATRX和P53核表达缺失,OLIG2阴性。AYA患者中H3及IDH野生型HGGs是一组异质性肿瘤。低级别肿瘤恶化也可能发生在这种亚型中,需进一步研究相关分子驱动因素(BRAF、p.V600E、FGFR、p53以及错配修复基因突变等)。与IDH野生型成人型HGGs相比,儿童型HGGs中MGMT启动子甲基化等标志物的预后相关性尚不明确。
AYA人群的局限性星形细胞胶质瘤主要有毛细胞型星形细胞瘤、具有毛样特征的高级别星形细胞瘤、多形性黄色星形细胞瘤、室管膜下巨细胞星形细胞瘤和脊索样胶质瘤。目前分类分级诊断依赖组织学特征。KIAA1549-BRAF融合(毛细胞型星形细胞瘤)和BRAF-p.V600E突变(多形性黄色星形细胞瘤)不仅有助于肿瘤诊断,还为靶向治疗提供指导。
对于大多数AYA胶质瘤,建议手术切除行组织病理诊断,除非MRI显示为稳定的、小的、无症状的、非增强肿瘤和位于功能区的深部肿瘤。对于NF1并发视神经通路胶质瘤或弥漫性内生桥脑胶质瘤患者等不建议行组织病理检测。所有胶质瘤均应当进行最大程度的安全切除。除减少肿瘤细胞外,足够的肿瘤组织对形态学评估、免疫组织化学染色和分子检测是必不可少的。对于AYA的CNS弥漫性胶质瘤,无论组织学分级如何,建议进行免疫组化(IDH1 R132H抗体)评估是否存在IDH突变,如果IDH阴性,则进行非典型IDH突变基因分型。如果存在IDH突变,应当进行1p/19q共缺失(诊断少突胶质细胞瘤)和ATRX染色(染色丢失诊断星形细胞谱系)进一步分析。AYA胶质瘤患者中,如果R132H免疫染色结果为阴性,建议对IDH1和IDH2进行测序,以检测不常见的IDH1和IDH2突变(图1)。
目前,IDH突变型胶质瘤患者的治疗仍依赖于预后标志物,如分级、患者体质状态、年龄、切除范围和神经系统症状。约25%AYA HGGs患者为IDH突变型。对于高危IDH突变肿瘤(WHO 3/4级、老年人、肿瘤残留、合并伴临床症状的病变)患者,初次肿瘤手术切除后,标准治疗通常包括放疗和替莫唑胺或PCV(丙卡嗪,洛莫司汀和长春新碱)的联合化疗。有研究表明,弥漫性胶质瘤患者中不仅存在IDH突变,还可能伴有PI3K-mTOR-AKT信号通路的改变。错配修复缺陷(mismatch repair deficiency,MMRD)与IDH突变型胶质瘤进行烷基化化疗有关。这类复发性肿瘤往往级别较高,治疗反应性较差,针对这一耐药途径的可靶向性仍在研究中,评估这些患者的突变负担和MMRD可能有助于打开临床试验或治疗途径。在新诊断的高级别IDH突变型胶质瘤或既往用烷基化化疗治疗的AYA复发胶质瘤患者中检测MMRD(免疫染色或测序)是有临床价值的。
在AYA人群中,由于具有成人型GBM分子特征的HGG在老年人中出现较少,因此IDH野生型HGG的分子检测方法略有不同。在中线IDH野生型HGG中,应通过H3 K27me3和H3.3 p. K27M免疫染色来评估H3 p.K27突变。如果H3K27发生改变,分子表征可能有助于进入临床试验。对共突变的评估,也可能为靶向治疗提供选择。目前,伴H3 p.k 27突变的儿童DMGs预后较差,与成人型GBM相似(中位生存期18.5个月)。
对半球HGGs,建议免疫组化检测H3 p.G34R。这种突变比成人型GBM或伴H3 k27改变的DMG预后更好,但比IDH突变的HGG预后更差(中位生存期36.2个月)。目前,H3 G34R突变型HGGs的治疗方法与成人型GBM相似,最大限度的安全切除联合放化疗(替莫唑胺)。H3p.G34R突变型HGGs也可能存在激活血小板衍生生长因子受体α(PDGFRA)突变,这可能对未来的治疗有影响。在确诊H3及IDH野生型HGGs中,分子检测将能够评估相关的拷贝数改变和染色体臂变化(7号染色体扩增和10号染色体缺失、EGFR扩增),以及非GBM的IDH野生型肿瘤(TERT启动子、BRAFV600E、MYCN、MMR、EGFR、PDGFRA、p53)中的其他相关突变。非GBM的IDH野生型胶质瘤AYA患者中,应当进行单核苷酸多态性(single nucleotide polymorphism,SNP)分析和/或RNA测序筛查其他突变。MGMT启动子甲基化是IDH野生型GBM预后预测生物标志物。对这些突变的识别可能为BRAF/MEK和FGFR抑制剂靶向治疗以及临床试验打开大门。
所有胶质瘤通常都要进行IDH1 R132H、p53和ATRX免疫组化染色。如果ATRX阳性,p53阴性,则确定1p/19q是否共缺失。如果IDH1 R132H阴性,通常在年轻患者中进行非典型IDH测序,或者根据临床病史进行测序。最后,在IDH突变型星形细胞瘤中,评估CDKN2A基因以进行分级。当IDH阴性,必须从分子水平区分GBM和儿童型弥漫性胶质瘤。在这种情况下,BRAF p.V600E可以用免疫组化检测,可能发生在IDH野生型LGGs 的AYA患者中的其他改变(FGFR1、FGFR2、MYB、MYBL1和BRAF)需要分子检测进行诊断(图2)。
神经元和神经胶质混合性肿瘤中,节细胞胶质瘤和胚胎发育不良性神经上皮肿瘤(dysembryoplastic neuroepithelial tumors,DNETs)是两种最常见的神经胶质瘤。其他胶质神经元肿瘤包括具有少突胶质细胞瘤样特征和簇状核弥漫性胶质神经元肿瘤、形成菊形团的胶质神经元肿瘤、乳头状胶质神经元肿瘤、粘液样胶质神经元肿瘤,弥漫性软脑膜胶质神经元肿瘤、节细胞瘤、多结节和空泡状神经元肿瘤以及Lhermitte-Duclos病。中枢神经细胞瘤归为神经元肿瘤。
胶质神经元肿瘤偶尔有潜在的靶向分子改变。在DNETs中,种系或FGFR1体细胞突变很常见,而BRAF p.V600E突变很罕见。节细胞胶质瘤中,BRAF p.V600E突变发生在10%-60%的病例中(取决于肿瘤位置)。玫瑰花形胶质神经元肿瘤可发生FGFR1突变,伴PIK3CA和NF1共突变。
WHO CNS5根据组织病理学、解剖学和分子特征将室管膜瘤分为10个亚组(图3)。由于分子特征数据尚不成熟,不同部位的室管膜瘤可以根据组织学特征分为2-3级。在WHO CNS5中,“间变性”一词已被删除,由于经典室管膜瘤的形态学变体(乳头状、透明细胞、伸展细胞)缺乏临床应用,已不再认为是室管膜瘤的亚型,而是被纳入组织学类型。
图3. 室管膜瘤诊断的年龄和基于解剖部位、组织学和分子特征的分类。
目前,室管膜瘤的治疗分层基于解剖位置、切除范围、分级和是否存在播散,而不是分子亚型。临床共识表明,在获得更多的试验数据之前,治疗应针对室管膜瘤的不同分子变体进行调整。图4展示AYA室管膜瘤患者分子检测法则。
幕上室管膜瘤(Supratentorial ependymoma,STE)WHO CNS5将STE划分为ZFTA(C11orf95)基因融合阳性和YAP1基因融合阳性两组;ZFTA基因融合阳性受累人群主要为AYA,YAP1基因融合阳性受累人群主要为婴幼儿。对于AYA的幕上室管膜瘤患者建议通过荧光原位杂交技术(FISH)、逆转录酶聚合酶链反应(RT-PCR)、下二代测序(NGS)或数字化基因检测技术进行ZFTA融合检测,如果不存在ZFTA融合,则应考虑其他诊断,如GBM伴室管膜分化或MN1融合或BCOR1融合神经上皮肿瘤。对于WHO 2级的STE伴ZFTA融合阳性患者,需通过FISH、SNP、甲基化分析进一步分析CDKN2A纯合性缺失。然而,对于STE的分子靶向治疗,还需要进一步的临床试验。
在WHO CNS5中,根据组蛋白H3 K27基化的整体水平,将后颅窝室管膜瘤分为PFA室管膜瘤(甲基化缺失和EZH抑制蛋白EZHIP过表达)、PFB室管膜瘤(甲基化保留)。大多数成人的后窝室管膜瘤属于PFB类,而绝大多数PFA室管膜瘤为8岁以下儿童(中位年龄为3岁)。在AYA人群中,与PFA室管膜瘤相比,PFB室管膜瘤更常见,预后更好。
由于其临床预后与经典脊髓室管膜瘤相当,WHO CNS5将黏液乳头状室管膜瘤列为2级。另外一种罕见脊髓室管膜瘤,伴有MYCN扩增。由于脊髓室管膜瘤是由形态学而不是分子学定义,因此,除非肿瘤表现为侵袭性或播散性,否则无需进行分子检测,在这种情况下,应通过FISH、SNP来检测MYCN扩增。
髓母细胞瘤(Medulloblastoma,MB)和胚胎性肿瘤中枢神经系统胚胎性肿瘤是高度恶性和低分化的神经上皮源性肿瘤。由于儿童和成人髓母细胞瘤有不同亚组和预后标志物,因此治疗策略和预后预测指标应有所区别。WHO CNS5中MB分子分型为:WNT活化型、SHH活化、TP53野生型、SHH活化和TP53突变型、非WNT/非SHH活化型。组织学分类为:经典型(classic,CL)、促结缔组织增生/结节型髓母细胞瘤(desmoplastic/nodular,DN)、具有广泛结节的髓母细胞瘤(medulloblastoma with extensive nodularity,MBEN)和大细胞/间变型髓母细胞瘤(large cell/anaplastic,LC/A)。大部分成人的MB属于SHH激活型,其次是Group 4和WNT激活型,Group 3 MB在成人中很罕见。在每个亚组中,进一步的转录和表观遗传变化已被证明有助于风险分层(表2)。
表2.髓母细胞瘤亚组的临床、分子信息以及危险分层。
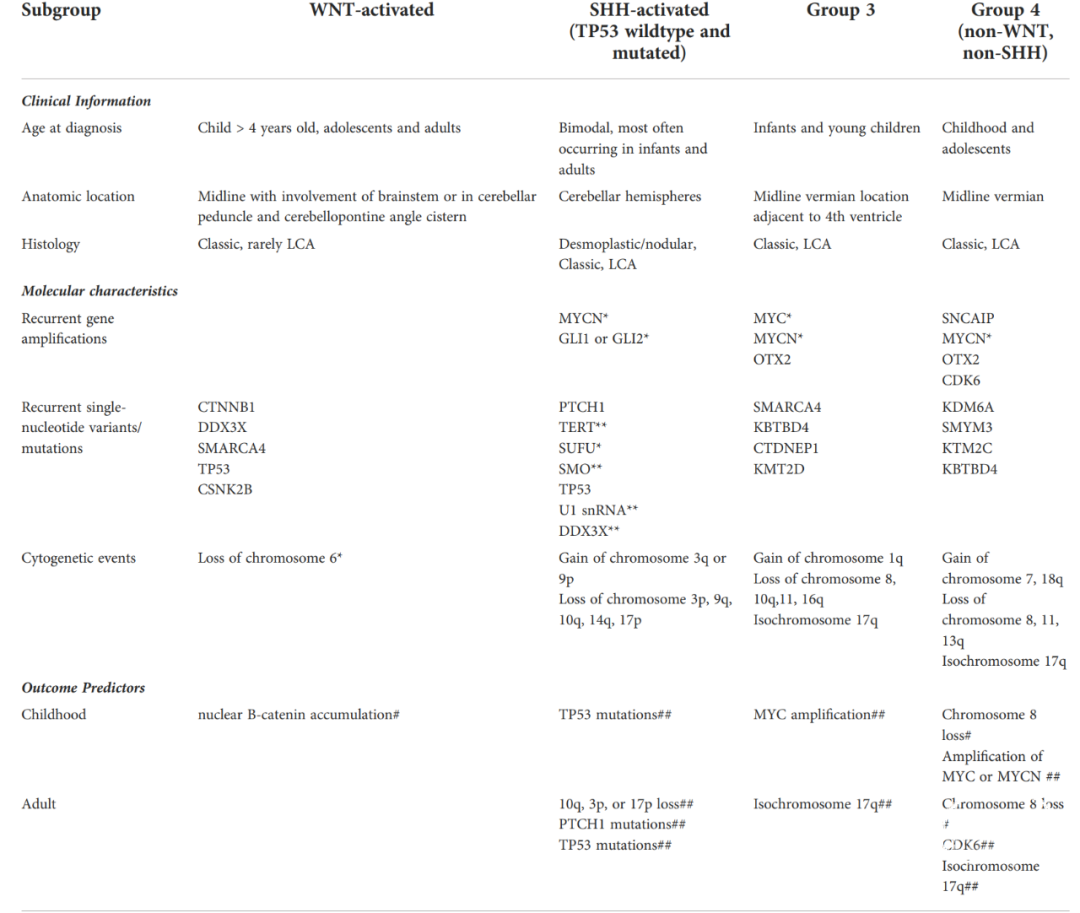
约15%-20%的成人MB属于WNT激活型;与儿童MB患者相比,成人MB不太可能存在6号染色体单倍体。大多数WNT激活型MB伴有CTNNB1基因的3号外显子突变,导致细胞质降解减少和β-Catenin核积累。在儿童中,β-Catenin核积累与良好的预后相关,而在成人中未发现这种预后价值。在成人中,SH激活型MB常见,而SH激活型MB中TP53野生亚型最常见,占70%。与儿童MB相比,P53突变不太可能与遗传癌症(Li-Fraumeni综合征)易感相关,并且在成人中P53阴性并不是预后标记物。TP53野生型SHH髓母细胞瘤富集TERT启动子突变,显示PTCH1中功能突变丢失或缺失,或拷贝数变化(10q丢失)。成人SHH髓母细胞瘤有频繁的上游通路改变(PTCH1和SMO突变),但很少有下游通路改变(SUFU,MYCN扩增)。10q缺失的存在是生存率差的一个强有力的预测因子。具有染色体3p缺失、17p缺失和PTCH1突变的SHH肿瘤预后较差。Group 3髓母细胞瘤主要见于婴儿和较大的儿童。多为男性患者,预后较差,尤其是伴MYC扩增者。其它的细胞遗传学特征,包括17q等臂染色体和8q染色体链。基因突变有SMARCA4,KBTBD4,CTDNEPI和KMT2D。Group 4髓母细胞瘤常出现细胞遗传学畸变,7或17q染色体扩增,8、11或17p染色体缺失以及17q等臂染色体。8号染色体丢失在儿童和成人中均证明与生存率增加有关,而其它儿童标志物,如11号染色体全丢失在成人中未发现与预后有关。伴有8号染色体丢失的儿童Group 4型MB与生存优势相关;MYCN和CDK6扩增以及PRDM6的过表达和组蛋白修饰基因KDM6A、AMYM3、KMT2C和KBTBD4的突变时,MYC或MYCN的扩增已证实与儿童型MB生存率低相关,但在成人中罕见。相反,CDK6几乎只在成人中发现,并与不良结果相关。髓母细胞瘤的诊断分为4个主要亚组是通过免疫组化和分子方法完成,后者是首选。免疫组化检测β-catenin可识别WNT亚组(β-catenin阳性),而SHH亚组、Group 3和Group 4的β-catenin阴性。在SHH亚组中,GAB1和细丝蛋白A阳性,但Group 3和Group 4呈阴性。目前没有可靠的免疫组化方法来区分Group 3和Group 4。儿童SHH激活型MB患者应接受遗传咨询以评估种系TP53和SHH通路突变(Gorlin综合征),无体细胞CTNNB1突变的WNT患者需要进行遗传咨询以评估APC测序。相比之下,成年SHH激活型MB患者通常不需要进行基因检测,因为老年患者很少有种系TP53突变,除非有其它临床或家族方面的问题。

声明:脑医汇旗下神外资讯、神介资讯、神内资讯、脑医咨询、Ai Brain 所发表内容之知识产权为脑医汇及主办方、原作者等相关权利人所有。
投稿邮箱:NAOYIHUI@163.com
未经许可,禁止进行转载、摘编、复制、裁切、录制等。经许可授权使用,亦须注明来源。欢迎转发、分享。









