排版 | AiBrain 编辑团队

慢性神经炎症是阿尔茨海默症(Alzheimer’s Disease,AD)的一个标志,表现为神经胶质增生、促炎因子水平升高和突触丢失1。
在小鼠模型和AD患者的大脑中,经典补体途径(Classic Complement Pathway,CCP)的激活导致神经元损伤和突触丢失,患者的大脑脑脊液的CCP因子异常升高,人类基因组学研究也支持CCP途径参与AD的发病机制2。
在AD小鼠模型中,药理学或遗传学抑制补体途径可改善神经退行性变和突触丢失3-4。在发育过程中,补体C1q与病原体、凋亡细胞结合,小胶质细胞吞噬补体标记的突触5。除了小胶质细胞外,星型胶质细胞也可以在发育过程中、成人大脑中和疾病中发挥突触清除作用6-8,然而,其发挥的突触清除作用的分子机制未知。
近日,来自美国Broad研究所Borislav Dejanovic团队及南加州Genentech公司Jesse E. Hanson团队合作在Nature Aging上发表了题为“Complement C1q-dependent excitatory and inhibitory synapse elimination by astrocytes and microglia in Alzheimer’s disease mouse models”的文章,通过深度蛋白质组学分析结合免疫染色,揭示了在AD小鼠模型中,小胶质细胞和星型胶质细胞共同通过C1q途径对兴奋性和性抑制性突触进行清除,并提示补体C1q或可成为治疗AD的靶点。

研究人员应用AD模型P301S转基因小鼠为研究对象,发现9个月的P301S小鼠的全脑脑容量和海马体积明显减少,而敲除C1q基因表达(P301S;C1qKO)的AD小鼠则几乎接近正常小鼠;相应地,P301S小鼠表现出低活动性(locomotor activity),而P301S;C1qKO小鼠活动正常(图1),提示敲除C1q对神经性退行有保护作用。此外,C1q缺失对P301S小鼠的Tau病理、胶质细胞增生、胶质细胞转录变化并没有显著影响。
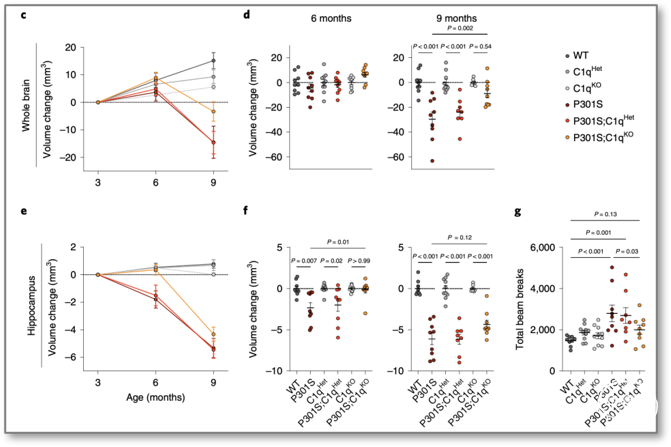
图1 C1q缺失可减少P301S小鼠的神经退行
(图源:Dejanovic B, et al., Nat Aging, 2022)
那么,C1q的缺失是如何起到神经退行的保护作用的呢?
研究人员通过多重串联质量标签(TMT)蛋白质组学分析,比较了不同转基因小鼠不同发育时间的突触蛋白组变化。发现6个月大的P301S突触中有108个下调和68个上调差异表达蛋白(Differentially Expressed,DE);在9个月时,有253种下调和434种上调DE(占总蛋白质的16.5%)。
相比之下,P301S;C1qKO小鼠海马的突触在6个月时仅显示17个下调和19个上调DE(占总蛋白质的0.5%),在9个月时有79个下调和224个上调DE(占总蛋白质的7%),C1q缺失不影响P301S大脑突触中的Tau水平(图 2)。
因此,这些结果表明:C1q缺失弱化了Tau病理学诱导的年龄依赖性变化;C1q的缺失并没有显著改变P301S大脑的转录组变化,暗示突触蛋白质组的变化很可能是由突触的局部蛋白质变化引起的。
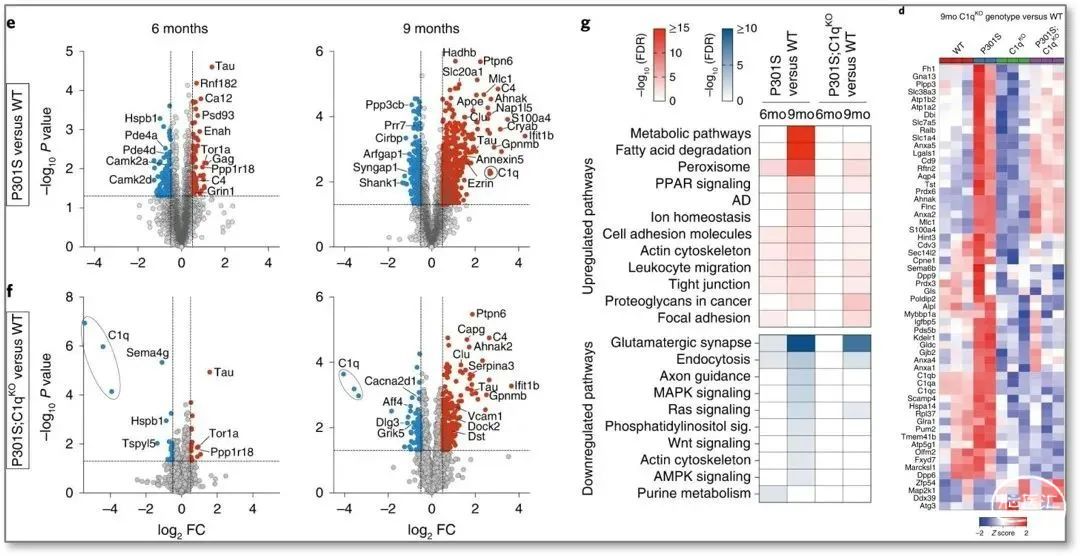
图2 C1q缺失减少P301S小鼠海马中突触蛋白的改变
(图源:Dejanovic B, et al., Nat Aging, 2022)
另外,研究人员注意到很多典型的星形胶质细胞特异性蛋白,如Aqp4、Mlc1和Slc1a4等,在9个月大的P301S突触中以C1q依赖性方式增加(图2d),随后利用pseudobulk single-cell RNA测序分析发现大多数上调的蛋白质主要由神经胶质细胞表达,且与是否表达C1q高度相关。
KEGG途径分析显示,P301S突触DE蛋白主要富集于“代谢途径”、“脂肪酸降解”、“过氧化物酶体”、“过氧化物酶体增殖物激活受体”信号通路,9个月大时“阿尔茨海默病”和“离子稳态”相关信号显著富集;然而,在P301S;C1qKO突触中,这些通路的富集相对较弱(图2g)。
进一步地,通过免疫电镜技术发现,在P301S小鼠的海马突触中,GFAP-Homer1接触显着增多,提示星形胶质细胞与兴奋性神经元的之间的联系增强。而在P301S;C1qKO小鼠则与正常小鼠无差异(图3),说明星形胶质细胞以C1q依赖的方式增加与突触的接触。
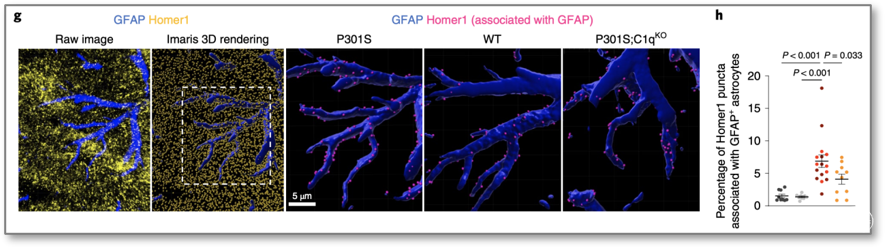
图3 C1q缺失使星胶与突触的接触减少
(图源:Dejanovic B, et al., Nat Aging, 2022)
有意思的是,在P301S小鼠的海马突触中升高的神经胶质蛋白,包括补体因子C1q和C4,星形胶质细胞标记蛋白MLC1和GFAP,小胶质细胞GPNMB和ANHAK 以及膜联蛋白是AD突触神经体中增加最多的蛋白质(图4a);研究人员在AD患者的脑脊液中也发现总C4浓度和加工(裂解和活化)C4浓度显着增加(图4b),提示CCP途径与AD进程中的神经退行密切相关。
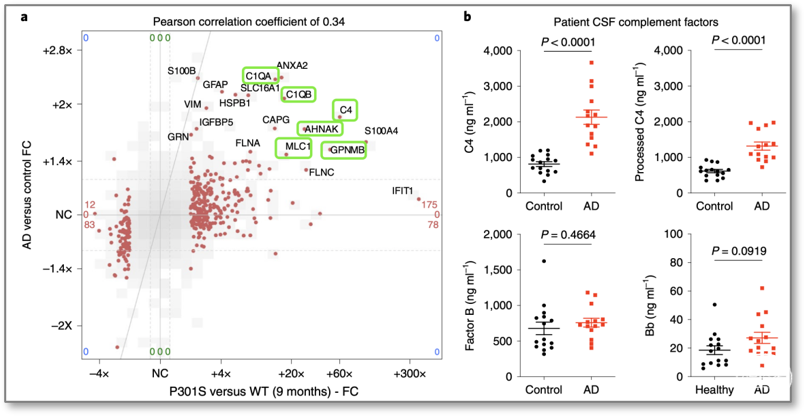
图4 AD患者突触中胶质蛋白表达增加,CSF中C4水平升高
(图源:Dejanovic B, et al., Nat Aging, 2022)
基于蛋白质组学、免疫电镜技术和免疫组织化学实验数据,研究人员假设在突触吞噬过程中星形胶质细胞可能以C1q依赖性方式与突触相互作用。通过共染星形胶质细胞GFAP+、小胶Iba1+、溶酶体Lamp1+、兴奋性突触后Homer1,抑制性突触gephyrin(图5),发现,P301S小鼠小胶溶酶体内的兴奋性突触数量远远多于P301S;C1qKO小鼠。
令人意外的是,P301S小鼠星形胶质细胞溶酶体内的兴奋性突触数量也多于P301S;C1qKO小鼠,说明星形胶质细胞和小胶质细胞以补体依赖的方式清除P301S小鼠的突触。
C3为C1q下游的重要补体组分。与野生型小鼠相比,P301S小鼠海马中C3+兴奋性突触后斑点(puncta)百分比显著增加;P301S;C1qKO小鼠较P301S小鼠C3+兴奋性突触后斑点数量显著减少,但总的C3 puncta百分数两者并没有显著差异,表明C1q的缺失特别影响C3在突触的积累;也进一步说明CCP促进星形胶质细胞和小胶质细胞消除突触。
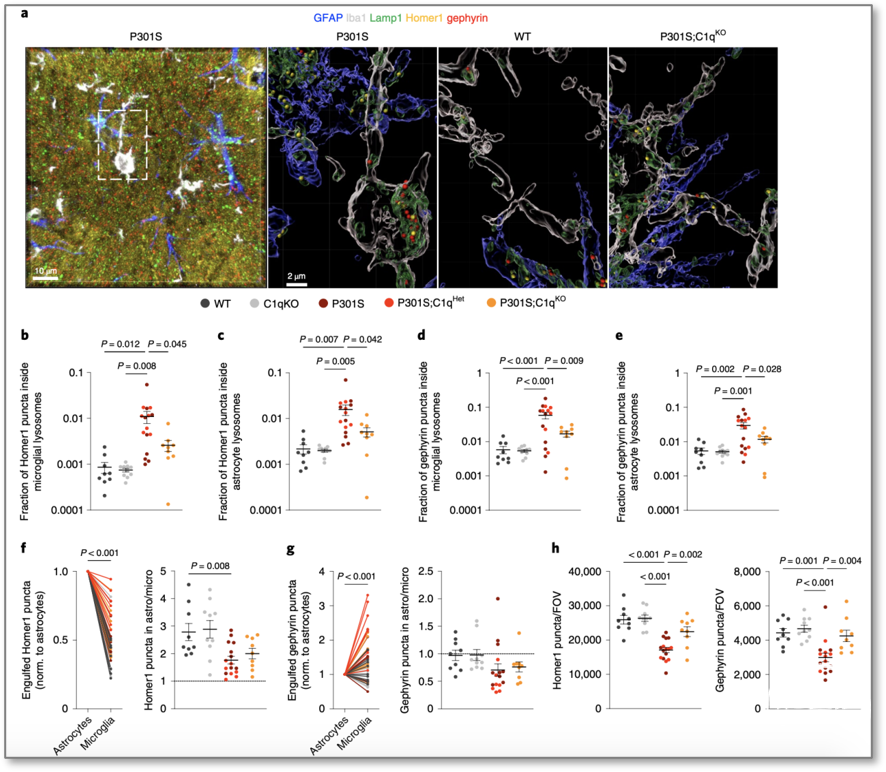
图5 星胶和小胶以补体依赖的方式清除P301S小鼠的突触
(图源:Dejanovic B, et al., Nat Aging, 2022)
那么,当星形胶质细胞(星胶)或小胶质细胞(小胶)的吞噬功能受损时,突触清除是如何进行的呢?
已知AD小鼠模型中小胶特异性TREM2缺失会抑制小胶质活化、向β-淀粉样(Aβ)斑块的迁移和吞噬活性。因此为了小胶功能障碍是否会影响星胶对突触清除的功能,研究人员分析了TREM2缺失在AD模型TauPS2APP小鼠中的影响,该小鼠同时包含了Aβ和Tau病理学特征。
同样地,利用免疫共染的方法发现,在TauPS2APP大脑中,与无斑块区域相比,小胶和星胶在斑块附近吞噬更多的突触(图 6);且与P301S小鼠相比,TREM2缺失使星胶溶酶体中兴奋性和抑制性突触的数量比小胶多。因此,当小胶吞噬功能受损时,星胶可代偿性地部分吞噬抑制性突触。
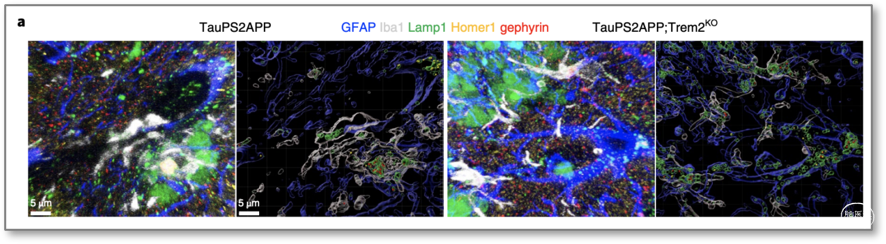
图6 Trem2缺失后斑块附近的突触清除
(图源:Dejanovic B, et al., Nat Aging, 2022)
文章结论与讨论,启发与展望
总而言之,研究人员对蛋白质组数据的深度解析,发现星形胶质细胞在补体依赖性兴奋性和抑制性突触清除中的新作用,实验还表明星形胶质细胞和小胶质细胞在病理生理过程中在突触吞噬中的伴随/协调作用。
然而,本研究是在固定的AD模型小鼠组织中进行免疫染色,因此无法动态性地区分两种神经胶质细胞是对突触进行在进行突触清除修剪(pruning)还是吞噬作用(cleaning)。进一步有关于神经胶质细胞与神经元功能的研究也有待验证。
首先,尽管研究表明C1q的缺失特别影响C1q下游重要补充组分C3在突触的积累,然而;是否存在特定的星形细胞受体可以直接检测到神经元上的补体沉积,或者是否可能存在间接性的补体依赖信号以触发突触周围星形细胞的局部激活,还需要进一步的实验证据。
其次,尽管本研究表明当小胶质细胞吞噬功能受损时,星形胶质细胞可代偿性地部分吞噬抑制性突触,提示星形胶质细胞与小胶质细胞在损伤组织重构过程中可能存在协同作用,然而,有研究表明,在多发硬化小鼠模型中,小胶质细胞而非星形胶质细胞通过补体通路消除突触9。
因此,星形胶质细胞和小胶质细胞有可能在突触处感知不同的补体分子。
原文链接:
https://www.nature.com/articles/s43587-022-00281-1
参考文献(上下滑动阅读) :



声明:脑医汇旗下神外资讯、神介资讯、脑医咨询、AiBrain所发表内容之知识产权为脑医汇及主办方、原作者等相关权利人所有。未经许可,禁止进行转载、摘编、复制、裁切、录制等。经许可授权使用,亦须注明来源。欢迎转发、分享。