病史简介
患者,男性,59岁,因“言语不清半月”入院。患者半月前无明显诱因下出现言语不清,伴有下肢麻木,无明显头痛头晕、无恶心呕吐、无肢体抽搐等不适。外院头颅MRI提示脑肿瘤可能。现患者为求进一步诊治来我院,门诊拟“脑肿瘤”收住入院。我院头颅MRI增强:左侧颞叶及脑室旁见多发结节状异常影。淋巴瘤或多发脑胶质瘤可能(图1)。
初步诊断:淋巴瘤或胶质瘤。

图1.术前头颅MRI 左侧颞叶及脑室旁见多发结节状异常影,等低T1(A)、T2/FLAIR高信号(B、C)、DWI高信号(F),周边见水肿信号,增强后见明显强化(D、E);左丘脑见结节状等低T1(A)、高T2/FLAIR信号(B、C)、DWI低信号(F),增强后未见强化(D、E),左侧脑室受压变细,局部中线右偏。
病理诊断
大体检查:(左丘脑)灰白灰红碎组织一堆,大小2×1×0.2cm,质软。
显微镜观察:肿瘤细胞在脑组织中浸润性生长(图2-A),细胞密度中等(图2-B),细胞轻度异型性,胞浆丰富、粉染,呈肥胖型星形细胞样(图2-C),未见明确核分裂像,可见小灶钙化(图2-D)。
免疫组化结果(图3):GFAP(+),Olig-2(+),IDH1R132H (-),BRAF V600E(-),P53(-),ATRX(存在),NF(+),Ki-67(2-5%+)。
分子检测结果:IDH 1/2外显子4野生型(Sanger测序法),不符合1p/19q杂合性共缺失(FISH检测),EGFR基因扩增阳性(FISH检测,图4-A),符合PTEN基因缺失(FISH检测,图4-B),TERT启动子C250T突变型(Sanger测序法),MGMT启动子甲基化(Sanger测序法)
最终诊断:(左丘脑立体定向活检)整合诊断:胶质母细胞瘤,IDH野生型。
组织学分类:弥漫性星形细胞瘤,肥胖细胞型,WHO分级:4级。
分子信息:IDH 1/2外显子4野生型(Sanger测序法),不符合1p/19q杂合性共缺失(FISH检测),EGFR基因扩增阳性(FISH检测,图4-A),符合PTEN基因缺失(FISH检测,图4-B),TERT启动子C250T突变型(Sanger测序法),MGMT启动子甲基化(Sanger测序法)
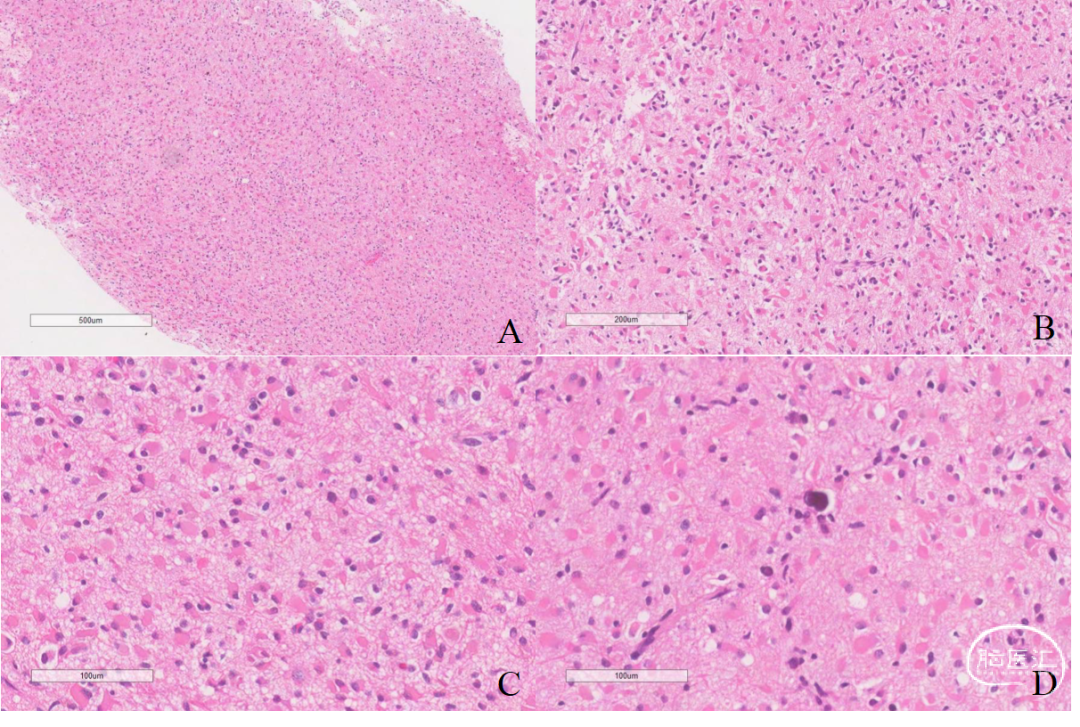
图2.肿瘤细胞浸润性生长(A),密度中等(B),细胞胞浆丰富、粉染,核偏位(C),局灶可见微钙化(D)。

图3.肿瘤细胞表达GFAP和Olig-2,IDH1 R132H阴性、P53个别肿瘤细胞核表达,ATRX存在,NF阳性,BRAF V600E阴性,Ki-67增殖指数约2%。
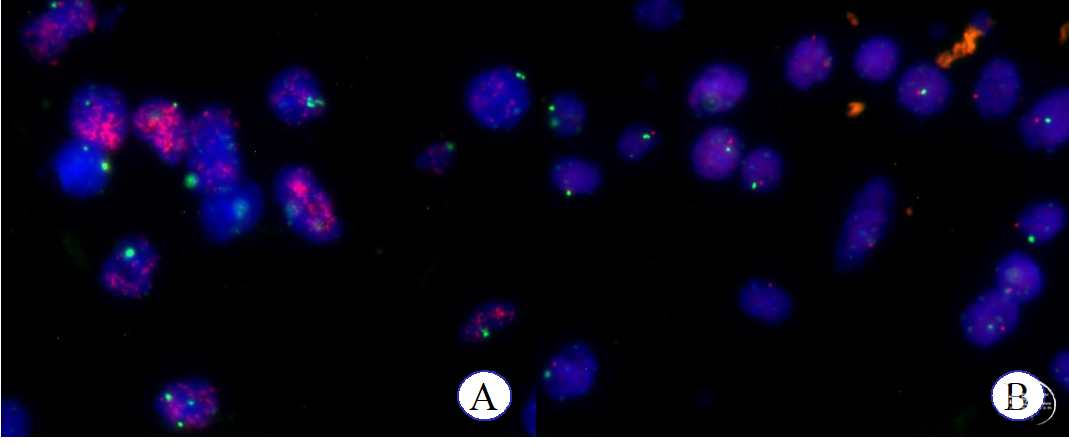
图4. FISH检测结果显示,EGFR基因扩增信号,红色荧光信号呈团簇状(A);PTEN基因缺失,双色荧光信号呈1红1绿(B)。讨论
2021年底,世界卫生组织(WHO)中枢神经系统(CNS)肿瘤分类第五版(以下简称WHO CNS5)正式面世,继2016版WHO CNS肿瘤分类开始将分子遗传学整合到肿瘤分类中之后,最新版蓝皮书在此基础上加入了更多分子遗传学内容,同时增加了一些以分子特征分型分级的肿瘤实体,以便优化临床中患者的治疗方案及预后判断[1]。
在WHO CNS5中,胶质母细胞瘤仅指IDH野生型,诊断标准除了组织学特征出现坏死和/或血管内皮增生外,对于组织学形态为2级或3级的IDH野生型星形细胞瘤,若具备以下分子事件一个及以上,即:表皮生长因子受体(EGFR)扩增、同时伴有整个7号染色体获得和整个10号染色体丢失(+7/-10)、端粒酶逆转录酶启动子(pTERT)突变,亦可直接诊断为胶质母细胞瘤,IDH野生型,CNS WHO 4级[2]。这些提示胶质母细胞瘤的分子标记物,是中枢神经系统肿瘤分类分子信息及实践方法联盟-非WHO官方组织(cIMPACT-NOW)为了对胶质瘤患者做更好的分层、预后判断和治疗选择,在回顾大量相关研究文献基础上筛出的,并于2018年以“cIMPACT-NOW 更新3”发表,当时称具有此分子特征的胶质瘤为“弥漫性星形胶质细胞瘤,IDH 野生型,具有胶质母细胞瘤的分子特征,WHO IV级”[3]。
EGFR扩增认为对胶质瘤侵袭性的临床行为预测具有极好的特异性,几乎不存在于其他具有惰性临床过程的胶质瘤中[4]。EGFR扩增是指基因的高水平拷贝数增加。仅低水平拷贝数增加,例如7号染色体三倍体,则不足以诊断EGFR基因扩增。同时,EGFR免疫组化检测虽敏感性高但特异性差,故不推荐临床中作为EGFR基因扩增的替代方法[5]。
+7/-10染色体特征同样具有极好的特异性,虽然报道有个别多形性黄色星形细胞瘤患者中也可出现,但BRAF V600E检测可帮助鉴别诊断[4]。有研究发现除了+7/-10之外,7p或7q获得以及10p或10q缺失的染色体改变与胶质瘤侵袭性行为、患者不良预后也高度相关[4,6]。但由于这部分胶质瘤比例较少且需要更多佐证,工作委员会决定依旧采用IDH野生型胶质母细胞瘤中最常见的染色体失衡,即+7/-10,作为组织学2级或3级的IDH野生型星形细胞瘤具有CNS WHO 4级生物学行为的分子标记物。
TERT启动子突变在IDH野生型胶质母细胞瘤中较为常见,并与肿瘤侵袭性行为密切相关。常与+7/-10或EGFR扩增相伴在IDH野生型弥漫性胶质瘤中出现,当然,这些标记物也可不完全重叠。cIMPACT-NOW更新3认为TERT启动子突变在没有+7/-10或EGFR扩增情况下,依然与侵袭性临床行为相关[6,7]。Tesileanu等的研究也证实了cIMPACT-NOW的结论,伴有胶质母细胞瘤分子特征的2/3级IDH野生型星形细胞瘤中位总生存期为23.8个月,与IDH野生型胶质母细胞瘤的19.2个月相近,并且仅pTERT启动子突变的2/3级IDH野生型星形细胞瘤总生存期也接近IDH野生型胶质母细胞瘤[8]。但近期Berzero等的研究结果不同,他们通过严格的病例筛选及神经病理专家复阅组织学后发现,伴有胶质母细胞瘤分子特征的2级IDH野生型星形细胞瘤中位总生存期为42个月,虽低于无胶质母细胞瘤分子特征的2级IDH野生型星形细胞瘤(中位总生存期57个月),但高于伴有胶质母细胞瘤分子特征的3级IDH野生型星形细胞瘤(中位总生存期17个月)。而且仅存在TERT启动子突变的2级IDH野生型星形细胞瘤的患者中位总生存期可达88个月,这不符合CNS WHO 4级胶质母细胞瘤的临床行为表现。认为组织学分级仍有预后意义[9,10]。这需要我们更多的临床实践及研究来明确。
胶质母细胞瘤分子标记物的应用对组织形态学的诊断有很好的补充,尤其在弥漫性星形细胞瘤活检标本、组织学形态显示不佳等情况下可减少标本取样所造成的局限性。除此之外,需要注意的是,此分子标记物的应用是在组织形态学基础上,弥漫性星形细胞瘤的诊断为前提。在一些其他IDH野生型胶质肿瘤中,例如多形性黄色星形细胞瘤、节细胞胶质瘤、伴有毛样特征的高级别星形细胞瘤和室管膜瘤等中也会出现TERT启动子突变[4,11,12]。另外,几乎所有少突胶质细胞瘤中存在TERT启动子突变,此时IDH突变及1p/19q共缺失检测是必须的,尤其在形态学特征不典型的活检标本中,需作鉴别诊断。
总之,分子遗传学在弥漫性胶质瘤的病理诊断中必不可少,完善的分子检测对于明确弥漫性胶质瘤的病理类型、CNS WHO分级、及更优化的临床治疗均非常重要。1.Wen, P.Y. and R.J. Packer, The 2021 WHO Classification of Tumors of the Central Nervous System: clinical implications. Neuro Oncol, 2021. 23(8): p. 1215-1217.
2.Louis, D.N., et al., The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol, 2021. 23(8): p. 1231-1251.3.Brat, D.J., et al., cIMPACT-NOW update 3: recommended diagnostic criteria for "Diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma, WHO grade IV". Acta Neuropathol, 2018. 136(5): p. 805-810.4.Stichel, D., et al., Distribution of EGFR amplification, combined chromosome 7 gain and chromosome 10 loss, and TERT promoter mutation in brain tumors and their potential for the reclassification of IDHwt astrocytoma to glioblastoma. Acta Neuropathol, 2018. 136(5): p. 793-803.5.Lee, M., S.Y. Kang, and Y.L. Suh, Genetic Alterations of Epidermal Growth Factor Receptor in Glioblastoma: The Usefulness of Immunohistochemistry. Appl Immunohistochem Mol Morphol, 2019. 27(8): p. 589-598.6.Aoki, K., et al., Prognostic relevance of genetic alterations in diffuse lower-grade gliomas. Neuro Oncol, 2018. 20(1): p. 66-77.7.Eckel-Passow, J.E., et al., Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N Engl J Med, 2015. 372(26): p. 2499-508.8.Tesileanu, C.M.S., et al., Survival of diffuse astrocytic glioma, IDH1/2 wildtype, with molecular features of glioblastoma, WHO grade IV: a confirmation of the cIMPACT-NOW criteria. Neuro Oncol, 2020. 22(4): p. 515-523.9.Berzero, G., et al., IDH-wildtype lower-grade diffuse gliomas: the importance of histological grade and molecular assessment for prognostic stratification. Neuro Oncol, 2021. 23(6): p. 955-966.10.Giannini, C. and F. Giangaspero, TERT promoter mutation: is it enough to call a WHO grade II astrocytoma IDH wild-type glioblastoma? Neuro Oncol, 2021. 23(6): p. 865-866.11.Koelsche, C., et al., Distribution of TERT promoter mutations in pediatric and adult tumors of the nervous system. Acta Neuropathol, 2013. 126(6): p. 907-15.12.Reinhardt, A., et al., Anaplastic astrocytoma with piloid features, a novel molecular class of IDH wildtype glioma with recurrent MAPK pathway, CDKN2A/B and ATRX alterations. Acta Neuropathol, 2018. 136(2): p. 273-291.(本文由浙二神外周刊原创,浙江大学医学院附属第二医院病理科许素素住院医师整理,许晶虹副主任医师审校,张建民主任终审)声明:脑医汇旗下神外资讯、神介资讯、脑医咨询、AiBrain所发表内容之知识产权为脑医汇及主办方、原作者等相关权利人所有。未经许可,禁止进行转载、摘编、复制、裁切、录制等。经许可授权使用,亦须注明来源。欢迎转发、分享。




