病史简介
患者,女,43岁,因“头痛3年余,加重1周”入院。
患者3年前无明显诱因出现头痛,无意识不清、无肢体抽搐等不适,未处理。1周前头痛较前明显加重,于当地医院就诊,行头颅MRI提示胼胝体前部占位。为求进一步诊治来我院急诊,行头颅CT:胼胝体膝部占位伴出血。
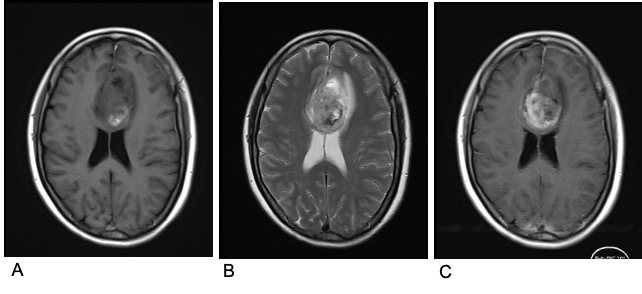
图1. 术前头颅增强MRI。A示T1WI信号混杂,等低信号为主,部分斑片状高信号,B示T2WI混杂高信号,夹杂部分高信号,C示增强后病灶不均匀强化。
第一次入院后完善相关检查。行开颅双侧胼胝体病损切除术。术中见肿瘤与脑组织和血管粘连紧密,血供丰富,仔细分离肿瘤于脑组织,于显微镜下大部分切除肿瘤。
病理诊断1
显微镜观察:肿瘤呈弥漫性生长,肿瘤细胞以梭形细胞为主,可见上皮样细胞成分及少量瘤巨细胞,具有多形性。肿瘤可见嗜酸性颗粒小体,血管可见淋巴袖套结构。未见核分裂像及坏死。
病理诊断:(胼胝体)多形性黄色星形细胞瘤,CNS WHO 2级。
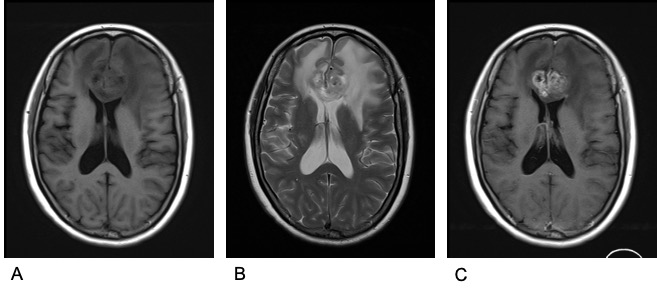
图2. 术前头颅增强MRI。A示T1WI低信号,B示T2WI混杂高信号,C示增强后病灶明显强化。
第二次入院后完善相关检查。行开颅双侧胼胝体病损切除术。术中见肿瘤与脑组织和血管粘连紧密,血供丰富,仔细分离肿瘤于脑组织,于显微镜下大部分切除肿瘤。
病理诊断2
显微镜观察:肿瘤呈弥漫性生长,部分区域可见黄色瘤样区域,上皮样细胞及梭形细胞成分均明显,见嗜酸性颗粒小体。可见灶性坏死,核分裂像<2.5个/mm²。
病理诊断:(胼胝体)多形性黄色星形细胞瘤,CNS WHO 2级。术后患者口服替莫唑胺,临床随访,半年后肿瘤再次复发。
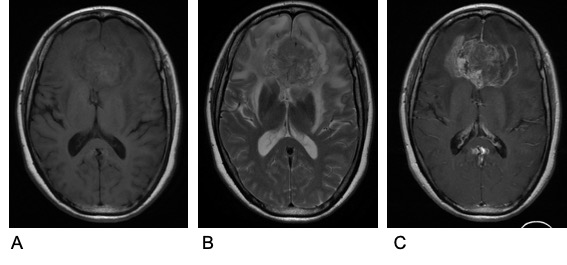
图3. 术前头颅增强MRI。A示T1WI等低信号,B示T2WI混杂高信号,C示增强后病灶不均匀花环状强化。第三次入院后完善相关检查。行开颅双侧胼胝体病损切除术。术中见肿瘤包绕侵犯大脑前动脉分支,于显微镜下次全切除肿瘤。
病理诊断3
镜下:肿瘤呈弥漫性浸润性生长,伴大片坏死,以上皮样细胞成分为主。核分裂易见,8-10个/mm²。病理诊断:(胼胝体)多形性黄色星形细胞瘤,CNS WHO 3级。
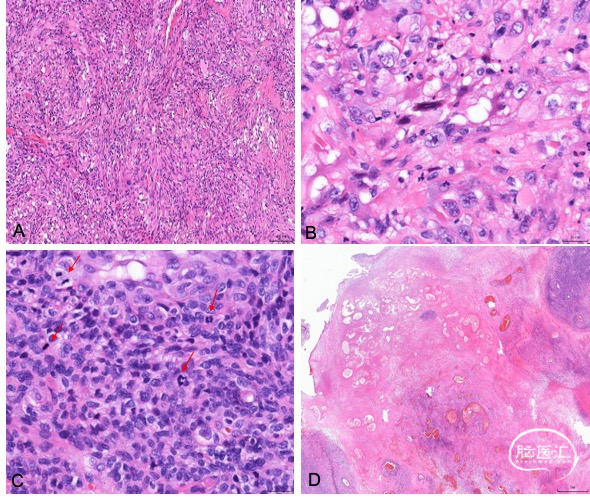
图4. 第三次肿瘤的显微镜下特点。肿瘤局部可见梭形细胞区域(A)及上皮样区域(B),显著增加的核分裂像(C,红色箭头),及大片坏死(D)。

图5. 第三次肿瘤免疫组化结果。GFAP肿瘤细胞局部表达(A),BRAF V600E弥漫表达(B),Ki-67增殖指数增高,可达40%(C)。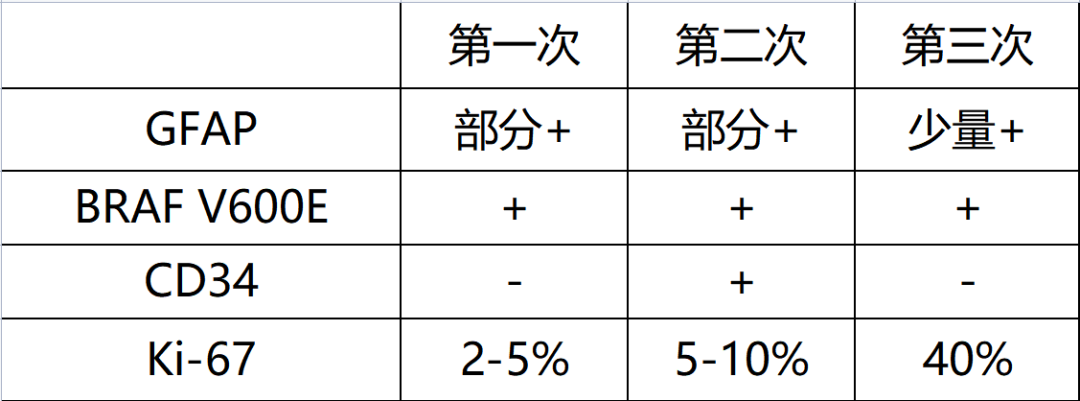
讨论
多形性黄色星形细胞瘤(Pleomorphic xanthoastrocytoma PXA)是一种好发于儿童和青少年的少见的星形细胞瘤。在2021年第5版中枢神经系统WHO分类中,将其归类于局限型星形细胞胶质瘤,包括CNS WHO 2级和3级。发病部位表浅,常累及软脑膜,98%病例发生在幕上,最常见于颞叶[1]。也有发生于小脑和脊髓的报道,并有两例儿童视网膜PXA的报道[2]。很多病例具有长期癫痫的病史,如果病灶位置较深(如脑干)或浸润范围广泛,则无法完全切除。
在原发性中枢神经系统肿瘤中,PXA不到0.3%。在男性和女性患者中发生率相同[3]。诊断时的平均年龄为26.3岁(中位年龄为20.5岁)。但老年患者也可能发生[4]。关于CNS WHO 2级肿瘤与CNS WHO 3级肿瘤的相对患病率数据很少。有报道31%的病例在首次诊断时即出现了间变性(23%的儿童患者和37%的成年患者)[5]。
在影像学上,PXA通常位于外周,多为囊性,累及大脑皮质和上覆的软脑膜。在CT上,肿瘤表现为可变(低密度、高密度或混合),具有强的、有时不均匀的对比增强。在MRI上,肿瘤的实体部分在T1加权图像上呈低信号或与灰质等信号,在T2加权图像上呈高信号或混合信号。相邻水肿通常不明显。
由于大多数肿瘤位置表浅,有推测PXA来源于软脑膜下星形细胞,且观察到软脑膜下星形细胞和PXA肿瘤细胞具有相同的超微特征。
大体上,PXA有时呈黄色(脂质化的原因),部分为囊性浅表皮质肿块(大体外观可不特异)。肿瘤可延伸到邻近的软脑膜。
在组织学上,肿瘤由梭形细胞、上皮样细胞和多核星形细胞混合组成,常常伴有脂滴结构(黄色瘤细胞)。可见到核内假包涵体、显著的核仁及淋巴袖套。嗜酸性颗粒小体是PXA特征性结构。软脑膜区域可以见到围绕单个肿瘤细胞的网状纤维沉积。CNS WHO 2级定义为核分裂像<2.5个/mm²,若核分裂像≥2.5个/mm²则为CNS WHO 3级。与2级相比,3级PXA表现为核分裂像活跃、多形性较少和浸润广泛的模式;坏死常见,但微血管增生不常见[4]。在3级的常见细胞学形态中,有小细胞、纤维性和上皮样/横纹肌样亚型[5]。
PXA通常表达GFAP和S100。S100常弥漫性阳性,GFAP至少局灶性阳性。大多数肿瘤CD34阳性,局部表达神经元标记物(synaptophysin、neurofilament、III类β-tubulin和MAP2)。
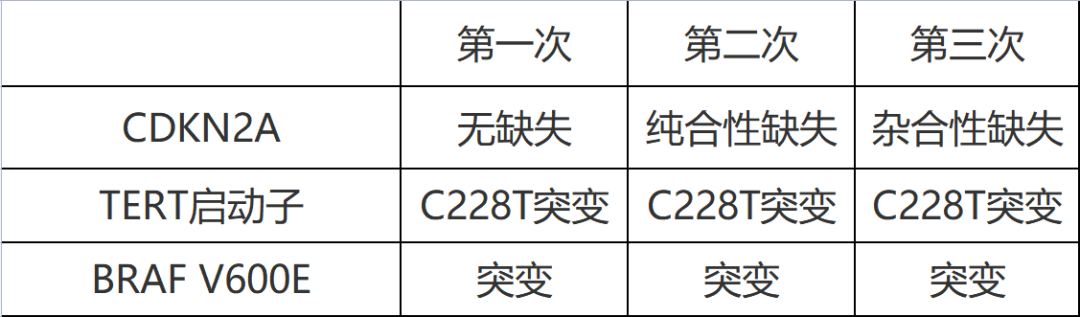
在分子病理学特征上,PXA通常携带MAPK通路成员的基因改变而导致此通路异常激活,最常见的是BRAF p.V600E突变,发生率在60%-80%;其余MAPK通路的基因改变有NTRK1, NTRK2, NTRK3, RAF1和NF1等。9p21处肿瘤抑制基因CDKN2A和/或CDKN2B的纯合性缺失在PXA的发生率也很高,有报道可高达94%[6,7]。此外,还发现不同比例的TERT启动子突变和(少见的)TERT基因扩增,这在3级PXA中较2级更常见[8]。而TP53突变在PXA中很少见[9]。BRAF p.V600E突变和CDKN2A/B的纯合性缺失是PXA的主要基因改变,与肿瘤的分级和预后无关。TERT启动子的改变认为与肿瘤更高的侵袭性有关,可作为间变的标志物。
在本例PXA的进展过程中,三次肿瘤切除标本的上皮样成分、核分裂像、坏死等特征逐次增多;GFAP表达下降,Ki-67增殖指数增加。BRAF V600E和TERT启动子突变均一致,但CDKN2A基因的缺失情况有不同。本例进展快,在术后一年内复发了两次,可能与TERT启动子的突变有关。从本例可以观察到PXA进展中形态学、免疫组化标记及分子特征的变化。
相对于其他局限型胶质瘤,PXA更易复发、扩散,并进展到更高级别的PXA[5]。早期干预和完全手术切除至关重要,2级PXA在大体全切除后需要随访观察,而3级PXA的CNS患者应接受术后治疗[10]。靶向治疗对于不能完整切除的患者来说是很重要的,即使肿瘤级别较低[11]。[1]Ida CM, Rodriguez FJ, Burger PC, et al. Pleomorphic Xanthoastrocytoma: Natural History and Long-Term Follow-Up. Brain Pathol 2015 Sep;25(5):575-86.[2]J O Zarate, R Sampaolesi. Pleomorphic xanthoastrocytoma of the retina. Am J Surg Pathol 1999 Jan;23(1):79-81.
[3]Quinn T, Gino Cioff, Haley Gittleman et al. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012-2016. Neuro Oncal. 2019 Nov 1; 21(Suppl 5): v1-v100.[4]C Giannini , B W Scheithauer, P C Burger et al. Pleomorphic xanthoastrocytoma: what do we really know about it?. Cancer 1999 May 1;85(9):2033-45.[5]Stephanie M Perkins , Nandita Mitra, Wan Fei et al. Patterns of care and outcomes of patients with pleomorphic xanthoastrocytoma: a SEER analysis. JNeurooncol. 2012 Oct;110(1):99-104.[6]Cristiane M Ida , Fausto J Rodriguez , Peter C Burger et al. Pleomorphic Xanthoastrocytoma: Natural History and Long-Term Follow-Up. Brain Pathol. 2015 Sep;25(5):575-86.[7]Joanna J Phillips , Henry Gong , Katharine Chen et al. The genetic landscape of anaplastic pleomorphic xanthoastrocytoma. Brain Pathol. 2019 Jan;29(1):85-96.[8]Christian Koelsche , Felix Sahm, David Capper et al. Distribution of TERT promoter mutations in pediatric and adult tumors of the nervous system. Acta Neuropathol. 2013 Dec;126(6):907-15.[9]C Giannini , D Hebrink, B W Scheithauer et al. Analysis of p53 mutation and expression in pleomorphic xanthoastrocytoma. Neurogenetics. 2001 Jul;3(3):159-62.[10]Rachael A Vaubel , Alissa A Caron , Seiji Yamada et al. Recurrent copy number alterations in low-grade and anaplastic pleomorphic xanthoastrocytoma with and without BRAF V600E mutation. Brain Pathol. 2018 Mar;28(2):172-182.[11]Michael Weller, Martin van den Bent , Jörg C Tonn et al. European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol. 2017 Jun;18(6):e315-e329.(本文由浙二神外周刊原创,浙江大学医学院附属第二医院病理科张慧主治医师整理,许晶虹副主任医师审校,张建民主任终审。)声明:脑医汇旗下神外资讯、神介资讯、脑医咨询所发表内容之知识产权为脑医汇及主办方、原作者等相关权利人所有。未经许可,禁止进行转载、摘编、复制、裁切、录制等。经许可授权使用,亦须注明来源。欢迎转发、分享。




