儿童视路胶质瘤是常见鞍区肿瘤,发病率仅次于颅咽管瘤。目前,国内外关于儿童视路胶质瘤的治疗无统一规范,是定期随访还是积极治疗;是放疗、化疗还是靶向治疗;若手术,肿瘤是全切、部分切除还是活检;众说纷纭,治疗效果大相径庭。据此,神外资讯采访了北京天坛医院小儿神经外科主任宫剑教授,作为全国最大的儿童视路胶质瘤诊疗中心,请他谈谈天坛诊疗规范,供全国同道借鉴。以下是访谈实录。
教授,主任医师,博士研究生导师,北京天坛医院小儿神经外科病区主任。中国医师协会神经外科医师分会小儿专家委员会副主任委员,中国医药教育协会小儿神经外科分会副主任委员。
主要研究方向:1. 儿童颅内肿瘤;2. 儿童颅脑先天性疾病。
目前主持科技部十三五、国家自然科学基金等多项课题,国内外专业杂志发表医学论著30余篇。
宫剑教授:
视路胶质瘤(Optic Pathway Glioma,OPG)是起源于视觉传导通路的星形细胞瘤,可累及视神经、视交叉、视束、视放射、下丘脑、第三脑室等多个解剖区域。儿童多见,60%发生于5岁之前的幼儿,男性多于女性,约占儿童颅内肿瘤的4-5%,儿童幕上肿瘤的15%[1-3]。视路胶质瘤多以水平眼震为首发症状,可伴视力减退、视野缺损;若累及下丘脑,表现为极度消瘦、性早熟;若堵塞室间孔或导水管,造成梗阻性脑积水,可出现头痛、呕吐等高颅压症状。视路胶质瘤依据解剖受累范围临床分型(Dodge分型,Ⅰ型占比25%、Ⅱ-Ⅲ型占比75%):Ⅰ型是肿瘤仅累及视神经;Ⅱ型是肿瘤侵犯视交叉;Ⅲ型是肿瘤侵犯下丘脑、突入第三脑室,可引发梗阻性脑积水[4]。视路胶质瘤经手术减压辅助放疗,5年生存率高达84.1%,预后良好[5]。视路胶质瘤组织病理以毛细胞型星形细胞瘤(WHOⅠ级)和毛黏液样星形细胞瘤(WHOⅡ级)多见[6];纤维型星形细胞瘤(WHOⅡ级)少见,高级别胶质瘤罕见[7-9]。儿童视路胶质瘤有何特点?如何与其它儿童鞍区肿瘤准确鉴别?宫剑教授:
首先要纠正一个误区,并不是儿童鞍区病变都是肿瘤,也并不是儿童鞍区肿瘤都是颅咽管瘤,若经验不足,将儿童鞍区病变当做颅咽管瘤加以根治性切除可能导致灾难性后果。儿童常见鞍区病变包括郎罕氏细胞增多症、垂体干淋巴炎、甲减继发垂体增生等,不需外科治疗[10, 11];儿童鞍区肿瘤最常见的是颅咽管瘤(33%-54%)、其次是视路胶质瘤(16%-20%)、生殖细胞肿瘤(7-14%),垂体瘤少见(3%)[12-16] [17-19]。各肿瘤治疗方法完全不同:颅咽管瘤需要根治性切除、鞍区生殖细胞瘤不需要手术而是通过化放疗加以治愈、视路胶质瘤需要部分切除配合放疗,因此,儿童鞍区肿瘤准确鉴别至关重要,一旦误诊误治,患儿可能致残致死。在此,对儿童鞍区肿瘤鉴别的关键点加以梳理,以期各位同道在临床实践中做出准确判断。
(1)颅咽管瘤:a. 主诉多为生长发育迟缓;b. 可出现视力减退、视野缺损,很少出现尿崩症;c. CT特征性影像为鞍区病变蛋壳样钙化。
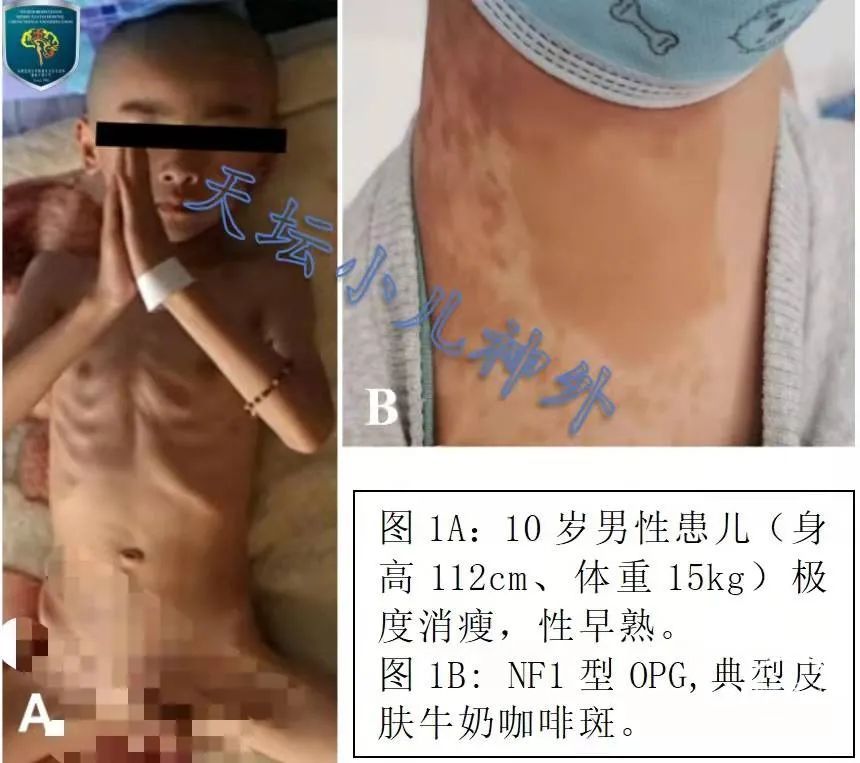
(2)视路胶质瘤:a. 水平眼震多为首发症状;b. 婴幼儿期多极度消瘦、恶液质表现,可伴有性早熟(图1A);c. CT呈低密度影、多不伴钙化;d. 若皮肤分布牛奶咖啡斑,则NF1型OPG诊断明确(图1B)。
(3)鞍区生殖细胞瘤:a. 多为青春期女性,首发症状为多饮多尿;b. CT呈团块状高密度影、多不伴钙化;c. 若血清HCG轻度升高则诊断明确[18]。
需要特别注意,儿童垂体腺瘤极为少见,75%为功能性垂体腺瘤[20],治疗目的是使垂体功能恢复正常。若手术导致垂体功能低下,发育停滞、不孕不育,则治疗不成功[20, 21]。因此,小儿神经外科医生对儿童鞍区病变的治疗应怀有敬畏之心,若诊断不明,宁可随访观察,也不应贸然手术,造成不可逆损伤(图2)。
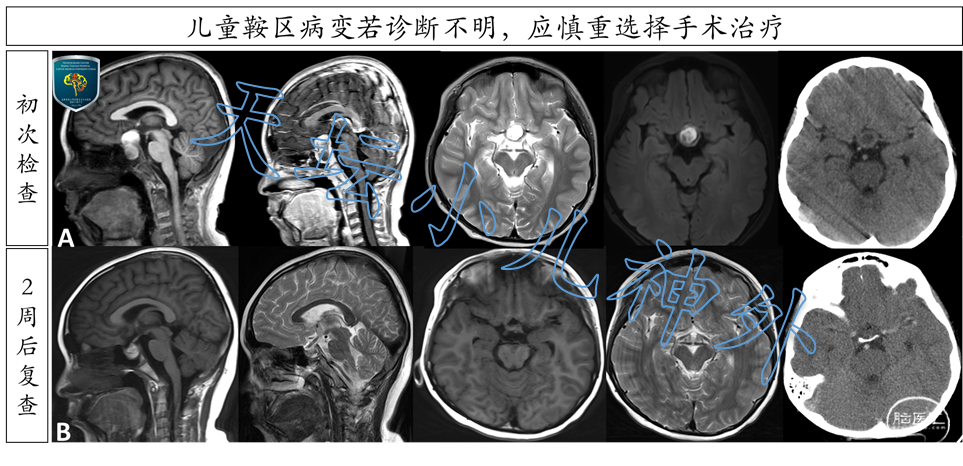
图2. 9岁男性患儿,主诉“头痛伴视物模糊1月余,逐步缓解”。头颅CT示鞍区混杂密度影,未见明显钙化;MRI示鞍区短T1长T2信号影,内部可见液平,外周环形强化(A排)。否认多饮多尿史,血清PRL轻度升高,FT4略降低,AFP(-)、HCG(-)。颅咽管瘤还是垂体瘤卒中实难决断,因此随访观察。两周后患儿头痛缓解、视力明显改善,鞍区病变显著缩小(B排),颅咽管瘤可除外。为什么谈视路胶质瘤(OPG),就必然提到神经纤维瘤病I型(NF1)?有何内在关联?首先谈一下神经纤维瘤病Ⅰ型(NF1)与Ⅱ型(NF2),二者均为常染色体显性遗传性疾病。NF1发病率约为33/10万,是定位于染色体17q11.2的NF1基因发生突变,临床表现为皮肤牛奶咖啡斑、皮下多发性神经纤维瘤等,损害程度与NF1基因突变类型有关,移码突变>剪切位点突变>错义突变[22]。最严重的是NF1基因大片段杂合缺失,表现为婴儿期面部畸形、智力低下、早发恶性肿瘤等[22, 23]。有报道国人23例NF1仅2例合并OPG[24],欧美人群约15%-20%的NF1合并OPG,发病率是国人的2-3倍[25]。NF2发病率约为1/25,000[26, 27],是定位于染色体22q12.2的NF2基因(抑癌基因)发生突变,90%-95%发生双侧听神经瘤、45%-77%发生脑膜瘤(均与NF2基因失活有关 )[28, 29]。
儿童视路胶质瘤合并NF1突变,即可明确诊断为NF1型OPG;反之,则诊断为散发型OPG。儿童视路胶质瘤为何要依据NF1突变分型呢?因为NF1型OPG的预后远好于散发型OPG。近2/3的NF1型OPG终身无进展,不需要手术干预;反之,近90%的散发型OPG持续进展,最终需要临床干预[30]。因此,当患儿初步诊断为OPG时,首先要判定是散发型还是NF1型,以确定进一步诊疗方案(表1)。需要指出,NF1型OPG欧美人多见,国外报道约30%的OPG患儿合并NF1突变[31];而我们曾对21例国人儿童OPG样本进行基因检测,仅1例存在NF1突变(4.8%),突变率远低于欧美人群。NF1型OPG多幼年起病、进展缓慢,常因皮肤咖啡斑就诊偶然发现颅内占位,高达70%的NF1型OPG没有临床症状,甚至存在肿瘤自然消退现象[32] (图3);若肿瘤稳定,10岁后进展的概率逐步降低,成年后再进展则极为罕见。总体上仅有1/3的NF1型OPG需要临床干预,预后较好[33]。反之,散发型OPG国人多见,全年龄段发病,因视力减退、头痛呕吐等发现颅内占位,病程短、进展快,90%需要临床干预(图4、图5)[30]。
表1:儿童视路胶质瘤NF1型与散发型之差异
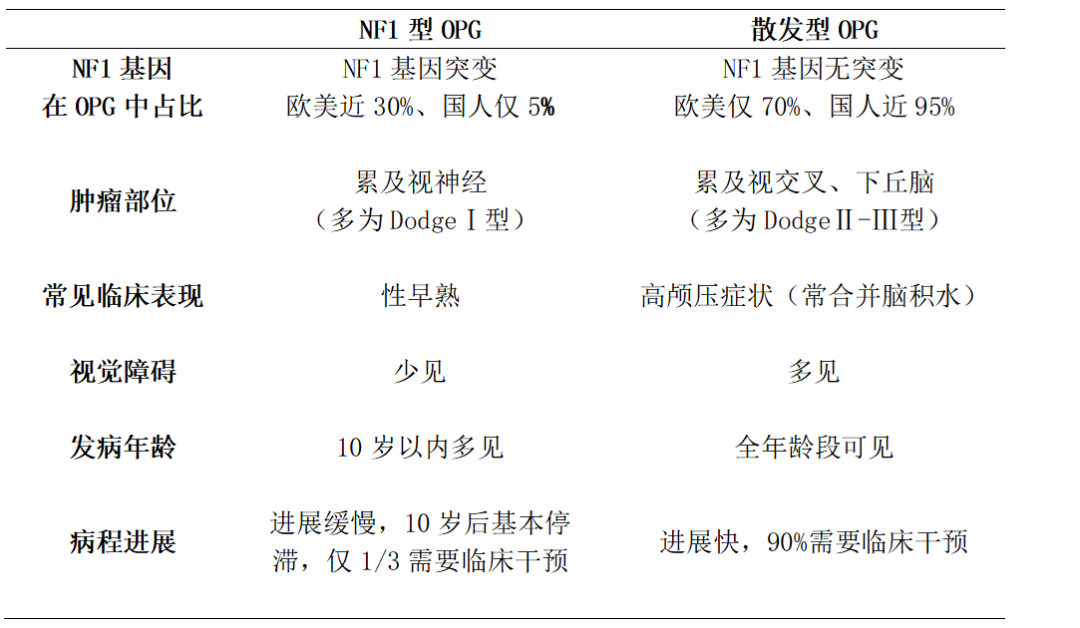
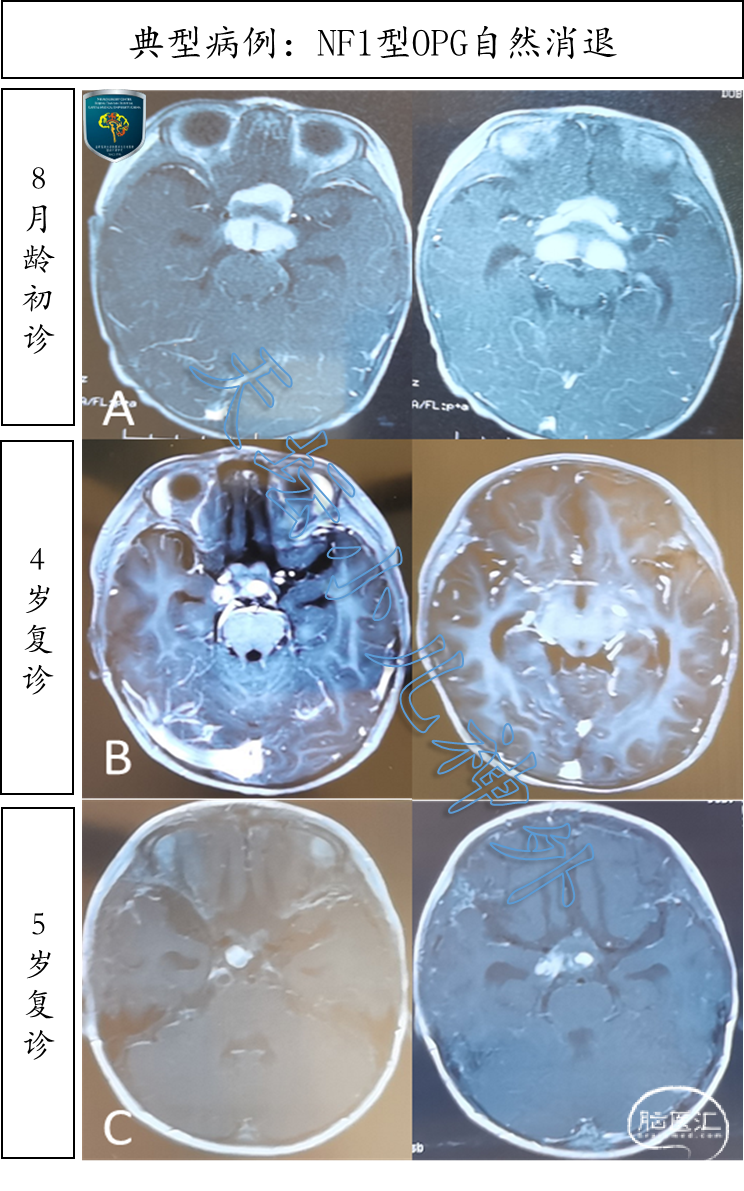
图3. 4岁女性患儿,因“新生儿黄疸”行颅脑MRI检查发现鞍区病变,外周血基因检测显示NF1突变,NF1型OPG诊断明确。A排:8月龄时可见视交叉、双侧视束占位;B排:4岁时瘤体明显缩小,提示肿瘤消退;C排:5岁时瘤体进一步缩小,自然消退趋势明显。
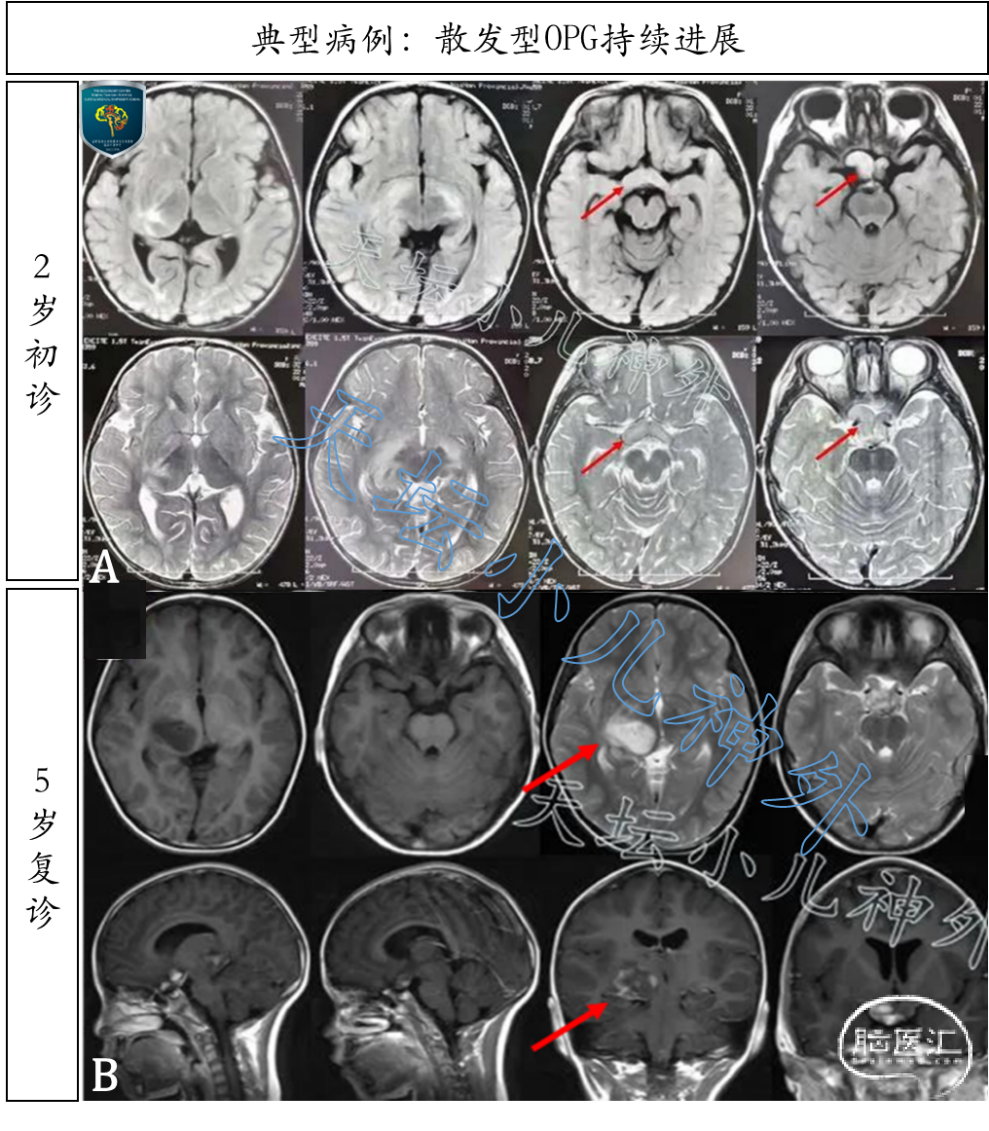
图4. 5岁女性患儿,主诉:双眼水平震颤3年,视力下降2年,头晕1月余。3年前发现视交叉、视束占位,外周血NF1基因突变(-),散发型OPG诊断明确。A排:2岁时发现视交叉、视束占位,因顾虑手术风险,选择动态观察;B排:5岁时右眼视力减退明显,MRI提示肿瘤进展,累及右侧丘脑、视放射。最终选择手术部分切除,病理回报:星形细胞瘤;免疫组化:IDH1(-),ATRX(+),P53(5%弱+),ki-67(约2-6%),H3K27me3(+),BRAFV600E(-);基因检测:NF1突变(-)。整合诊断:儿童型弥漫性低级别胶质瘤,NOS型。术后放疗效果好,视力较前改善明显。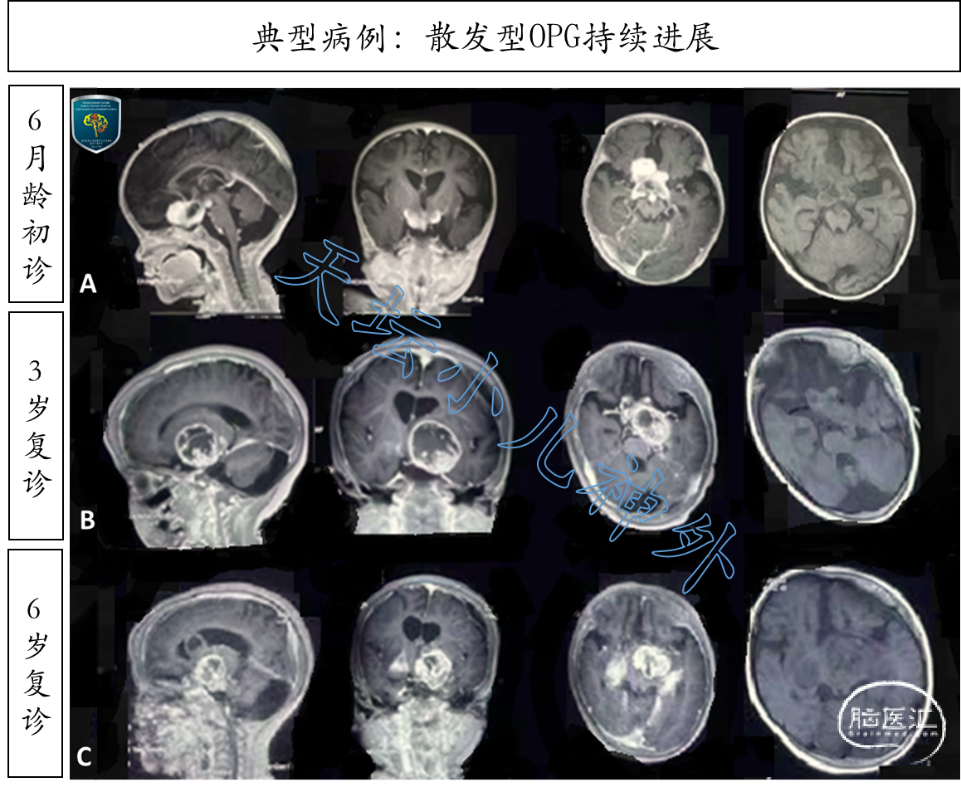
图5. 6岁男性患儿,6月龄时因头围增大发现鞍上占位,因顾虑手术风险,选择动态观察。A排: 6月龄显示鞍区占位;B排:3岁显示肿瘤明显增大,双侧视力减退明显;C排:6岁显示肿瘤持续进展,突入侧脑室并向对侧视束播撒,双眼仅存在光感。家长终于下决心手术,行肿瘤部分切除,病理回报:毛细胞型星形细胞瘤;免疫组化:IDH1(-),ATRX(部分+),P53(偶见+),ki-67(约1-3%),BRAFV600E(-);基因检测:BRAF突变(+),NF1突变(-);整合诊断:毛细胞型星形细胞瘤,伴BRAF突变。后续放疗,进行中。WHO CNS肿瘤分类新版变化较大,OPG在分类上该如何归属呢?
宫剑教授:
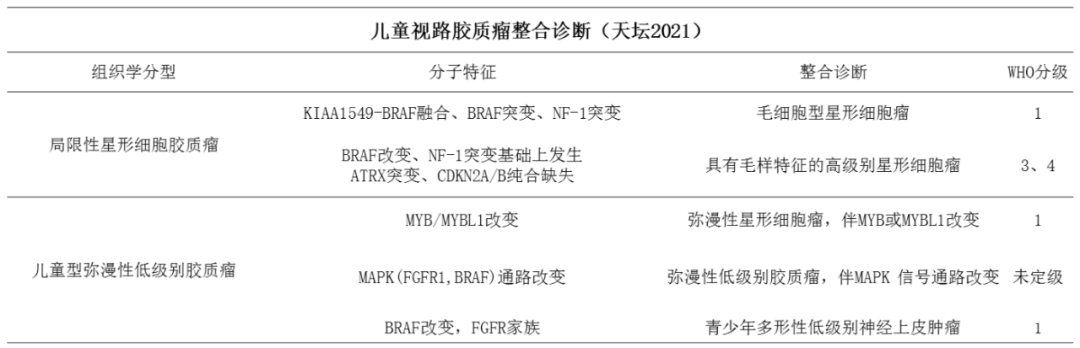
无论新旧版,都未对儿童视路胶质瘤进行单独分类。旧版(2016,第四版)以组织病理为基础,新版(2021,第五版)则强调基因变异并结合组织病理、免疫组化予以整合。迄今为止,国内外均未对儿童视路胶质瘤的分类进行梳理,据此,我们归纳出儿童视路胶质瘤(OPG)整合诊断(天坛2021),并期望在实践中不断完善(表2)。依据2016版分类,视路胶质瘤多属于毛细胞型星形细胞瘤(PA,WHOⅠ级)或毛黏液样型星形细胞瘤(PMA,WHO Ⅱ级),高级别胶质瘤仅占2%[34],胶质母细胞瘤极为罕见,全球仅见3例报道[7-9](图6)。
依据2021版分类,视路胶质瘤归为儿童型弥漫性低级别胶质瘤(DLGG)与局限性星形细胞胶质瘤(CAS)。前者包括(1)弥漫性星形细胞瘤,伴MYB或MYBL1改变;(2)弥漫性低级别胶质瘤,伴MAPK 信号通路改变;(3)青少年多形性低级别神经上皮肿瘤;后者包括(4)毛细胞型星形细胞瘤;(5)具有毛样特征的高级别星形细胞瘤[35]。
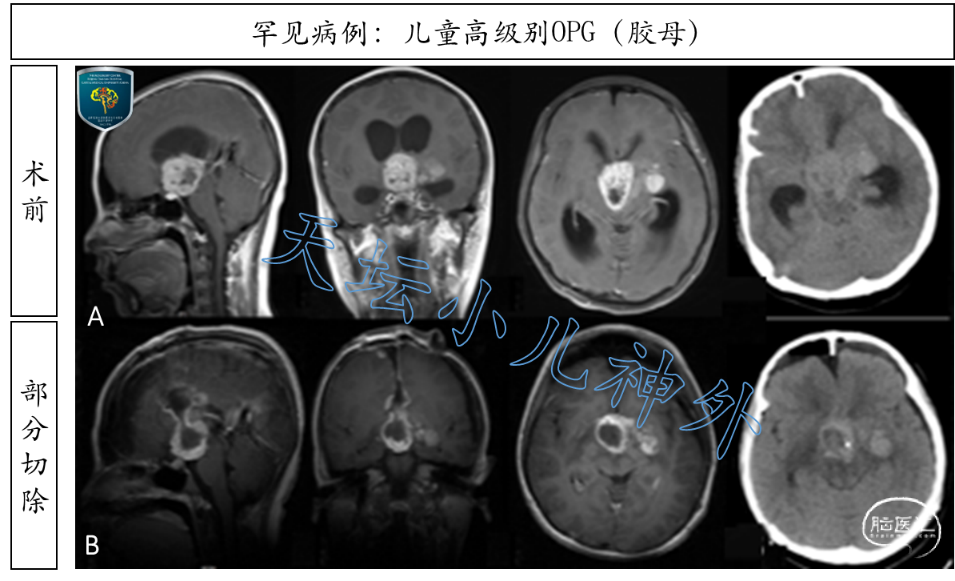
图6. 12岁女性患儿,主诉:视力减退一年,头痛加重1月。头颅CT/MRI显示:鞍上、视交叉、左侧视束、视放射占位,行肿瘤部分切除。病理回报:胶质母细胞瘤(Ki-67:30%);免疫组化:GFAP(+),Olig-2(散在+),SMA(血管阳性),S100(+),INI-1(+);基因未检测。整合诊断:儿童型弥漫性高级别胶质瘤,NOS型,术后4个月死亡。
研究显示,儿童低级别胶质瘤存在的基因突变主要与促分裂素原活化蛋白激酶(MAPK)信号传导通路上调的基因相关,如BRAF2突变或融合,FGFR1突变或重构,NF1突变以及NTRK基因家族融合等[36],70%以上的毛细胞型星形细胞瘤可观察到KIAA1549-BRAF基因融合[37, 38]。反之,ATRX、CDKN2A、TP53突变的儿童低级别胶质瘤预后较差[39],BRAFV600E突变合并CDKN2A/B纯合缺失可能导致低级别胶质瘤恶性转化[39, 40]。因此,利用分子特征对儿童低级别胶质瘤进行危险分层,有助于临床决策:合并NF1突变、KIAA1549-BRAF融合、CRAF突变的可归为低风险;合并BRAFV600E突变、IDH1突变、FGFR1突变的可归为中风险;合并H3K27M突变或BRAFV600E突变合并CDKN2A/B纯合缺失的归为高风险[40]。表2:儿童视路胶质瘤整合诊断(天坛2021)
备注:
*毛细胞型星形细胞瘤/毛黏液样型星形细胞瘤,若发生KIAA1549-BRAF融合、BRAF突变、NF1突变,在新版分类中归属为毛细胞型星形细胞瘤(1级)。
**毛细胞型星形细胞瘤/毛黏液样型星形细胞瘤若发生ATRX突变、CDKN2A/B纯合缺失,则提示恶性转化,在新版分类中归属为具有毛样特征的高级别星形细胞瘤(3、4级)。
迄今为止,国内外针对儿童视路胶质瘤均缺乏规范化治疗方案,何时治疗?如何治疗?是否需要手术?如何手术?术后是放疗、化疗、还是靶向治疗?治疗效果如何?既缺乏大宗病例总结、长时间随访,也缺乏令人信服的治疗效果与诊疗规范。作为全国最大的儿童视路胶质瘤诊疗中心,我们正着手进行梳理,有义务尽早推出天坛的数据与同行共享,造福广大患儿。
目前多数学者认为,对于偶然发现的视路胶质瘤,若无临床症状,应进行外周血检测,判定是散发型还是NF1型OPG。前者近90%患儿发生病情进展,需要临床干预,应坚持逐年复查,及时治疗;后者可长期随诊,10岁后肿瘤进展概率逐年减小,复查间隔可延长至1次/3-5年,仅1/3患者最终需要临床干预[33, 41, 42]。若出现临床症状,应积极手术治疗。我们与众多学者意见一致,OPG的手术治疗不可替代[43, 44] 。手术目的:①明确组织病理及分子特征,对指导后续治疗具有决定性作用;②充分减轻瘤负荷,尽量切断瘤内供血,使肿瘤从富血供向乏血供转化,提高放疗敏感性[45];③打通脑脊液循环,缓解高颅压,降低后续治疗风险。具体而言,对于局限于一侧眶内DodgeⅠ型OPG,若瘤体持续进展且存在颅内侵犯风险时,应及时请眼科医生评估是否行根治性切除以阻断肿瘤向颅内蔓延[46];若肿瘤累及视交叉、下丘脑(DodgeⅡ-Ⅲ型)出现相应症状,应由神经外科医生主导,采取部分切除,且要避免视路与下丘脑的损伤。
Jenkin认为,OPG手术部分切除后,应积极放疗,治疗效果好于手术后单纯观察的患者[43]。Awdeh报道,手术配合放疗对患者视力的恢复明显好于单纯放疗的患者。因此,放疗前瘤体充分内减压,视力改善效果明显[45]。放疗作为治疗OPG的传统方法,能够有效地控制肿瘤的进展(总剂量40~56Gy)。接受放疗的OPG患儿10年生存率高达80%~90%,视力、视野改善率达到30%~55%,明显好于化疗[47]。我国采用3岁以上即可接受放疗的标准,5-7岁以上散发型OPG,放疗是术后首选治疗[48]。化疗作为传统治疗方式,用于OPG的治疗至今已有近50年的历史。化疗更适用于轻症患儿,卡铂联合长春新碱(CV方案)治疗初诊和复发的OPG有效率分别为66.67%与50%[49],化疗对视力、视野的改善作用有限,部分患者化疗效果不佳,需要手术补救,总体治疗效果逊于手术治疗及放疗(表3)[43, 44, 50]。
表3:世界主要治疗中心儿童视路胶质瘤疗效对比[44, 51-55]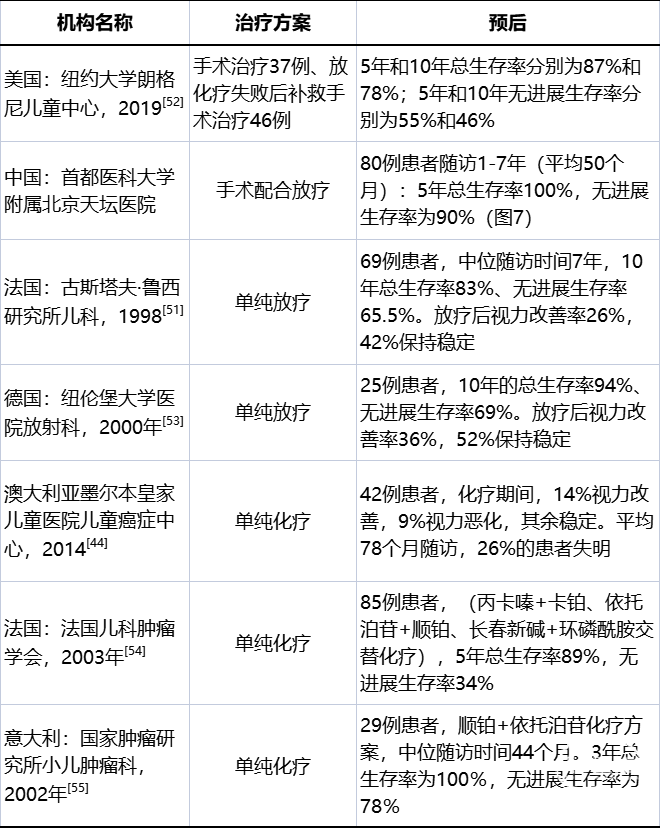
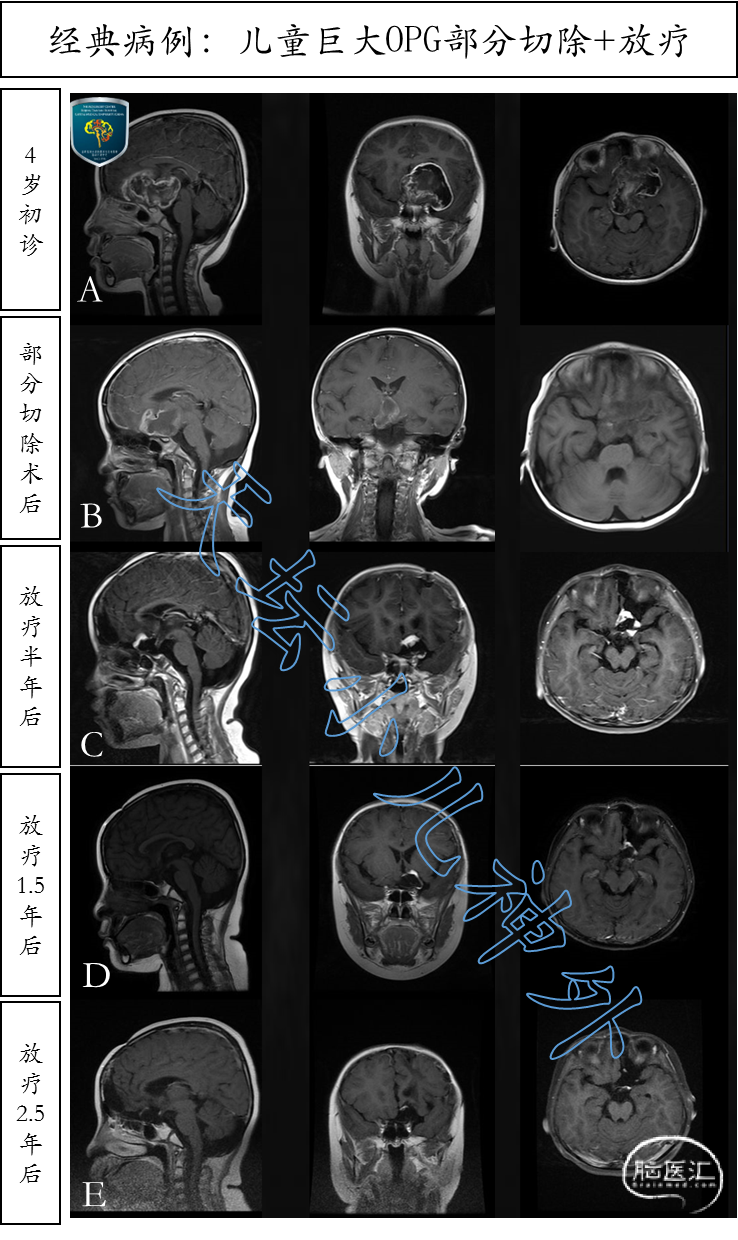
图7. 4岁女性患儿巨大视路胶质瘤,部分切除配合放疗,视力恢复好、激素水平正常,肿瘤基本消失,正常学习生活(图8)。A排: 4岁初诊时MRI显示鞍上、鞍旁巨大占位;B排:肿瘤部分切除,病理提示:毛细胞型星形细胞瘤,术后及时放疗;C排:放疗后半年肿瘤大部消失;D排:放疗后一年半肿瘤已不明显;E排:放疗后两年半肿瘤基本消失,疗效满意。
如前所述,众多学者认为,OPG的手术治疗不可替代,手术切除配合放疗是最佳选择。目前争议最大的是肿瘤切除程度,存在两个极端:有的学者顾虑手术风险,谨慎采用活检配合放疗[5];有的学者则激进的提出根治性切除,从而避免后续放化疗[51]。前者,事实上OPG根据临床与影像学特征基本可以明确诊断,活检并不是放化疗的前提[55],单纯活检不能减轻瘤负荷,配合放疗效果不佳[45]。而后者,则坚决反对。我们知道,视路胶质瘤起源于视觉传导通路、毗邻下丘脑,对肿瘤进行根治性切除,势必造成视觉传导通路的中断,这种医源性的损伤是难以接受的。同时,根治性切除必将造成严重的下丘脑损伤,本可以治愈的低级别胶质瘤因为手术导致昏迷甚至死亡,应严格避免。因此,我们认为儿童视路胶质瘤,部分切除配合放疗是最佳选择。B:若采用部分切除,切除范围可否量化?
儿童视路胶质瘤的手术,天坛小儿神外历经20年的改进与传承,究竟切除到什么程度,始终没有量化标准,口口相传的是“适可而止”,该如何领会呢?
如前所述,视路胶质瘤部分切除的目的:1. 明确病理;2. 充分减轻瘤负荷;3. 打通脑脊液循环。但是,明确病理可以通过活检实现,打通脑脊液循环不如脑室-腹腔分流更可靠。因此,手术的关键在于第2点:充分减轻瘤负荷,尽量切断瘤内血供,保证后续放疗效果[45]。若切除过少,术后残余肿瘤易发生卒中,严重者危及生命;若切除过多,易导致下丘脑损伤,严重者同样危及生命(图9)。“适可而止”该如何拿捏呢?笔者体会:
1. 视路胶质瘤的手术同样要遵循“中线肿瘤,中线入路”的原则,尽量避免侧方入路,造成视觉传导纤维的损伤。
2. 依据肿瘤生长方向,若选择前纵裂入路,下丘脑损伤轻,应注意视路的保护;若选择胼胝体-穹隆间入路,视路损伤轻,应注意下丘脑的保护。
3. 手术以减轻瘤负荷为目的,沿中线纵行切开肿瘤包膜,严格进行瘤内操作、囊内减压,避免游离肿瘤边缘造成视觉传导通路与下丘脑的损伤。要直视下操作,避免盲刮、盲拽,过度牵拉。
4. 视路胶质瘤存在高度的异质性,若呈胶冻样、血供不丰富,应尽量吸除,充分囊内减压;若血供丰富,瘤内富含成簇小分支动脉,应确切电凝止血,既防止残余肿瘤卒中,又尽可能使肿瘤由富血供向乏血供转化,提高对后续放疗的敏感性[2, 56, 57];若瘤体硬韧、钙化明显,可采用超吸(CUSA)瘤内减压,但切记保持包膜完整,避免突破包膜与基底池沟通,造成视交叉或下丘脑的损伤。
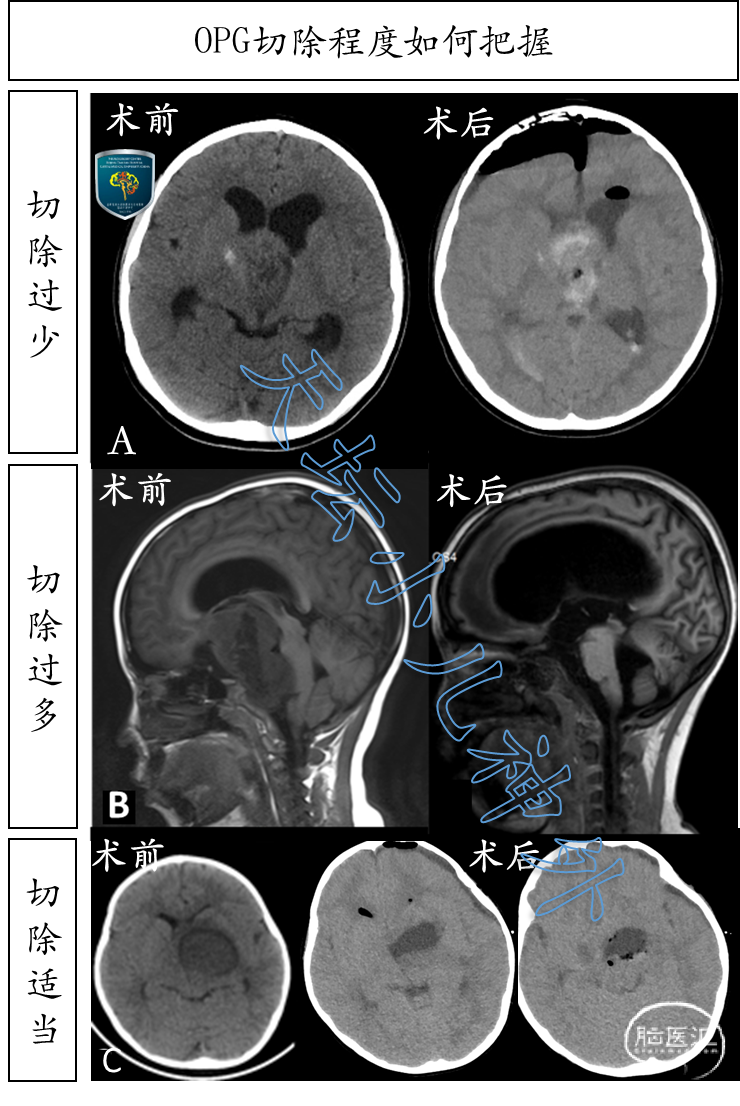
图9. A排:肿瘤切除过少,残余肿瘤卒中,紧急二次开颅清除;B排:肿瘤根治性切除,术后深昏迷,去脑强直,预后差;C排:肿瘤切除恰到好处,效果好。
C:NF1型OPG术后能否放疗:
NF1型OPG术后可否放疗目前存在争议。有学者认为放疗可导致儿童认知功能减退、内分泌功能障碍,甚至诱发新生肿瘤及恶性转化[13]。作为国内最大的儿童视路胶质瘤治疗中心,我们认为OPG的治疗是以挽救视力与保护生命为首要前提,术后选择放疗还是化疗,应该是哪个有效就选择哪个。事实证明,放疗优于化疗,特别是对视力改善,优势明显 [7, 33, 51]。至于放疗导致内分泌功能障碍,我们知道,累及下丘脑的肿瘤本身可导致内分泌功能障碍,进而诱发认知功能障碍[5, 58],哪些是放疗引起的,值得进一步探讨[59]。NF1患者随访过程,3%新生颅内肿瘤[25],若把新生肿瘤都归于放疗,未免牵强,需要更多病例加以证实。因此NF1型OPG患者并非放疗禁忌症[13]。
D:OPG合并梗阻性脑积水该如何处理?
视路胶质瘤多位于中线部位,易发生梗阻性脑积水(约30%)[2]。因此,在切除肿瘤前,通过脑室-腹腔分流缓解脑积水、解除高颅压,安全可靠(图10)。有学者提出通过切除肿瘤,打通脑脊液循环而避免分流手术,初衷很好,但临床可行性差。我们知道,视路胶质瘤的手术原则是部分切除肿瘤,明确病理、减轻瘤负荷,即使手术一时打通了脑脊液循环(解除肿瘤对室间孔、导水管上口的阻塞,图11),残余肿瘤的术后肿胀很容易再次阻塞脑脊液循环。再加上手术残渣、凝血块、止血材料等因素的影响及后续放疗过程中脑组织顺应性改变,术后脑积水再发率极高。我们回顾了近3年天坛小儿神外80例儿童视路胶质瘤,合并梗阻性脑积水40例,21例术前分流,顺利进行手术及后续放化疗,无脑积水再发;19例术前未分流,术后放化疗期间脑积水再发11例(57.9%),5例(23.8%)急诊行脑室-腹腔分流加以抢救,辅助治疗被打断,效果不佳(图12)。在此我们明确告诫同行,不要试图通过切除视路胶质瘤缓解梗阻性脑积水,疗效极不确切,脑积水再发率高,潜藏巨大风险:轻则打断后续治疗,重则突发脑疝、呼吸停止,危及患儿生命,我们都是有深刻教训的。因此,若OPG合并梗阻性脑积水,先行脑室-腹腔分流确切解除高颅压,为后续治疗全程提供安全保障,值得推广。
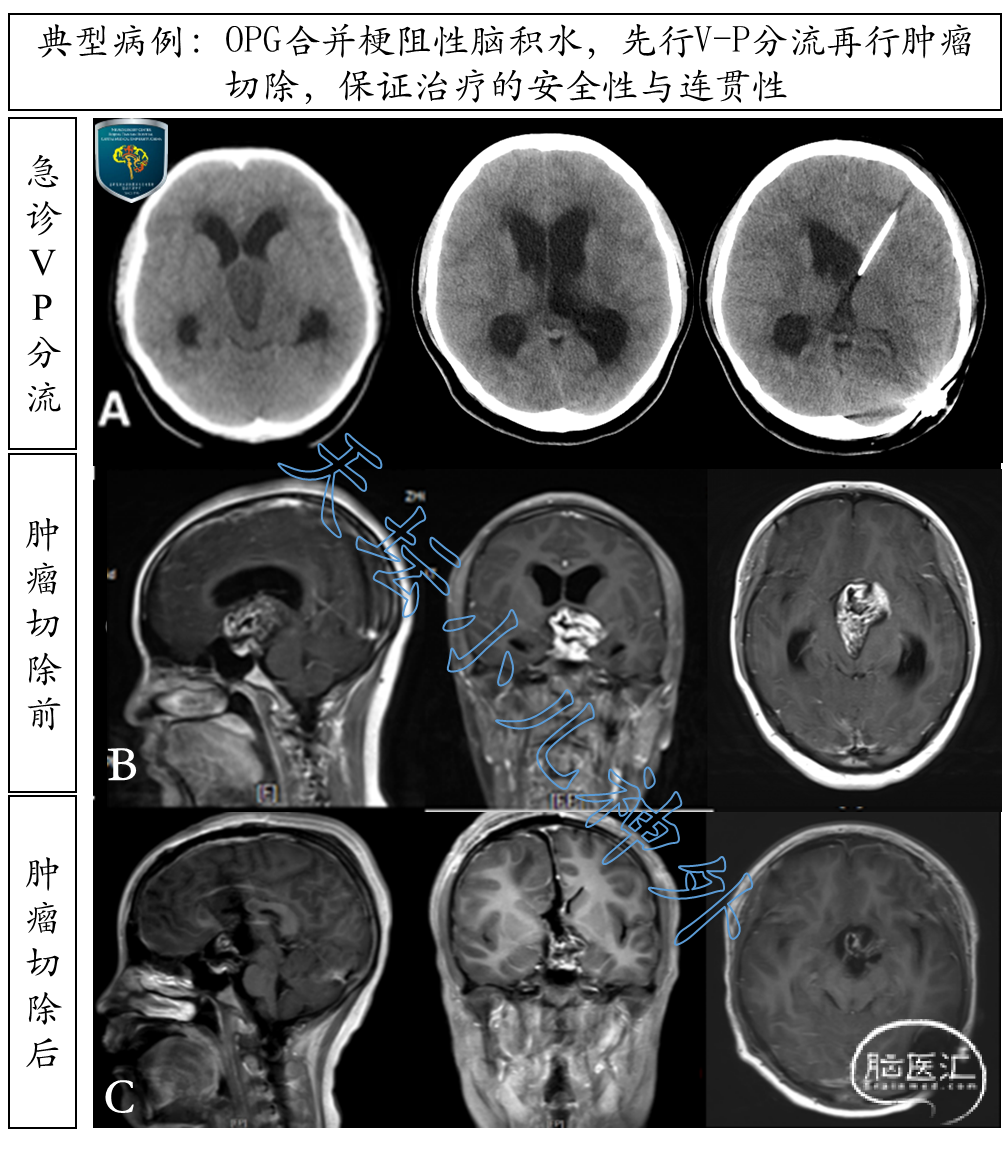
图10. 15岁女性患儿,主诉:左眼视力减退2月,头痛加重伴呕吐一周。头颅CT/MRI显示:视路胶质瘤,梗阻性脑积水(A排、B排)。急行左侧脑室-腹腔分流术缓解高颅压,患儿状态明显改善。继而行肿瘤大部切除术(C排),术后恢复好,左眼视力有所改善,顺利转入放疗。整个治疗过程连贯有序,一气呵成。
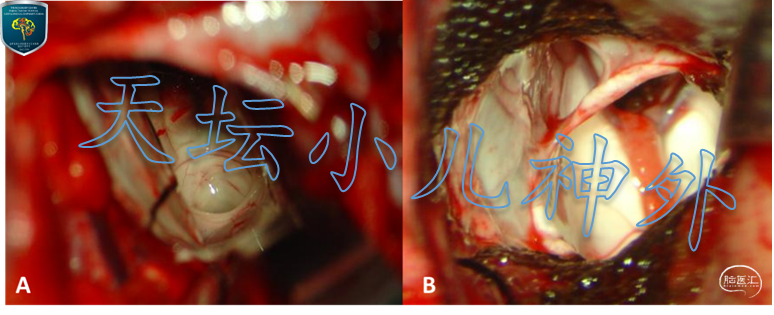
图11. 术中照片:A. 暴露导水管上口;B. 暴露右侧室间孔。
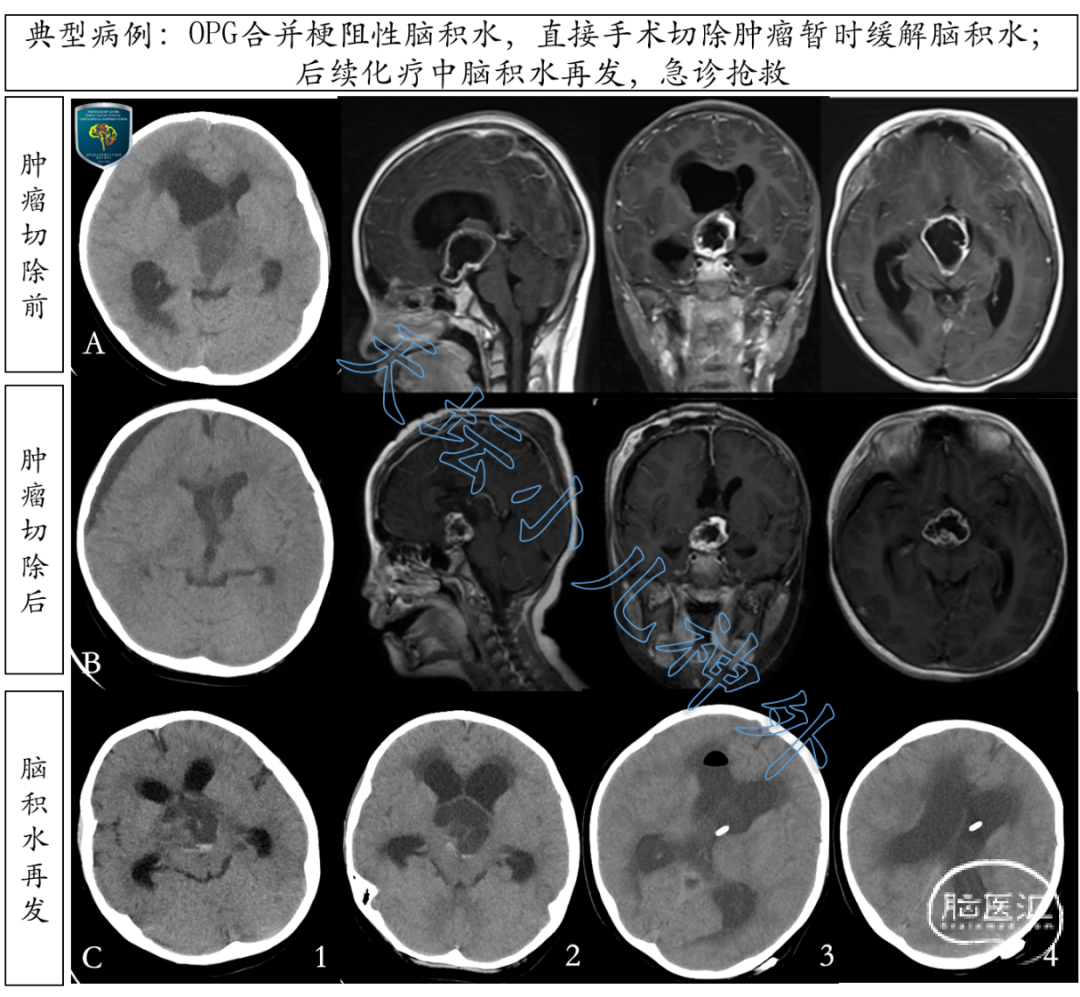
图12. 6岁男性患儿,主诉:间断性头痛呕吐2月余,左眼内斜视2周。头颅CT/MRI显示:视路胶质瘤,梗阻性脑积水(A排)。未先行分流,直接肿瘤切除,脑积水暂时缓解(B排)。后续治疗中,脑积水再发,病情危重(C1、C2),急行左侧脑室-腹腔分流术挽救生命(C3、C4),辅助治疗中断,效果不佳。
E:OPG肿瘤播散,治疗效果如何?
儿童视路胶质瘤多为组织学良性表现,但少数毛细胞型星形细胞瘤具有很强的侵袭性,约3-12%会出现播散转移[60-62],多发生于颅内或椎管内软膜下腔[63]。若OPG已出现播散转移,仍可通过部分切除配合全脑全脊髓放疗,以及口服替莫唑胺(TMZ)等方案综合治疗,患者的临床症状和影像学表现多会改善,部分患者可以治愈[62, 64, 65]。
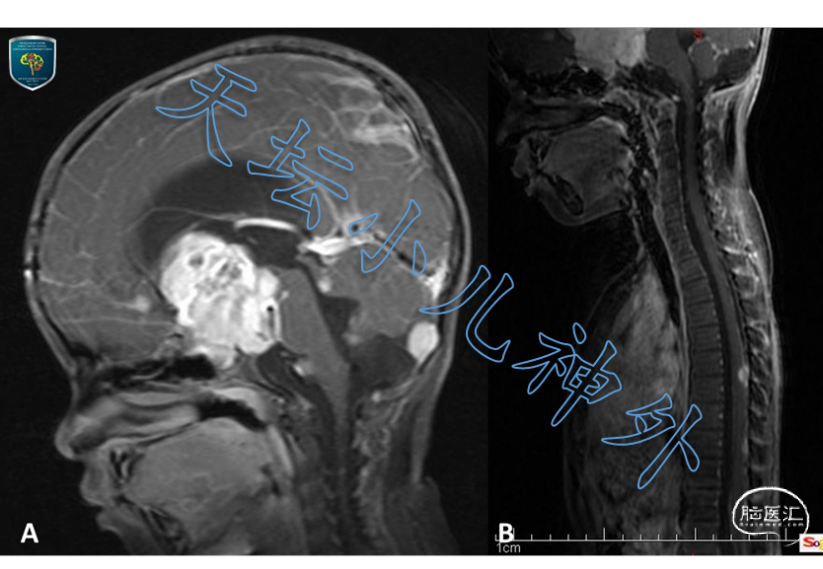
图12. 儿童视路胶质瘤,脑干、小脑、脊髓多发软膜下播散转移。
多数儿童低级别胶质瘤(LGG)与MAPK通路中相关分子上调有关,出现某些基因突变的OPG可试行靶向药物治疗[66]:达拉菲尼、维莫菲尼等BRAF抑制剂,对合并BRAFV600E突变的难治性OPG有效[67];但若应用于BRAF-KIAA1546融合或BRAF野生型OPG中,反而会加速肿瘤进展[68]。新一代BRAF抑制剂PLX8394目前还处于临床试验阶段[69],MEK抑制剂曲美替尼对一些难治性OPG有一定疗效[70],司美替尼在BRAF基因变异或NF1相关的LGG均有效,该药近期已获得美国FDA批准,可用于治疗>2岁NF1型OPG[71]。mTOR抑制剂依维莫司对复发进展性NF1型OPG疗效显著[57],抗血管生成剂贝伐单抗可使肿瘤缩小、改善患儿的视力[72]。FGFR、ALK、ROS1以及NTRK等突变在儿童OPG中较为少见,相关靶向治疗仍处于探索阶段[66]。
需要指出,各类靶向药物属于补充治疗。儿童视路胶质瘤的首选依然是减瘤手术加放疗,对此,广大同道与家长应该清醒认识。
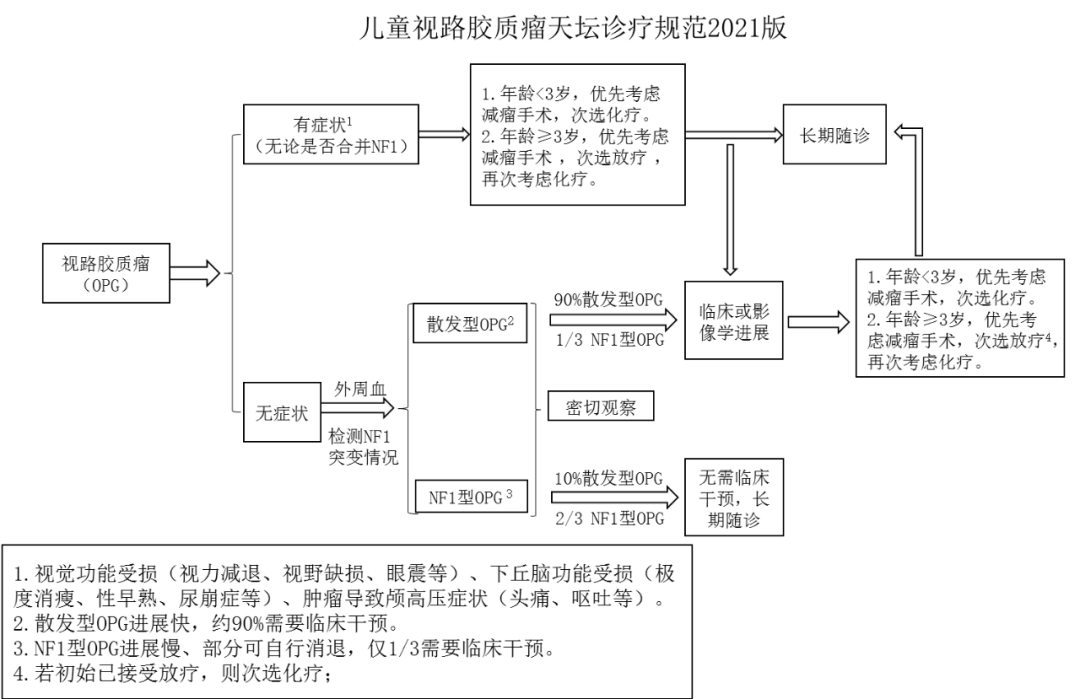
据此,我们结合大量临床实践,制定出儿童视路胶质瘤(OPG)天坛诊疗规范(2021):1. 儿童视路胶质瘤合并梗阻性脑积水,若出现头痛、呕吐等症状,应先行脑室-腹腔分流术缓解高颅压,为后续治疗提供安全保障。
2. 儿童视路胶质瘤若临床症状明显,应首选手术治疗。手术策略是肿瘤部分切除,明确病理,减轻瘤负荷,将肿瘤从富血供向乏血供转化而增加放疗敏感性。手术应注意视路与下丘脑的保护,禁忌根治性切除。
3. 术后若条件允许,应尽早(一月内)行放疗、次选化疗,肿瘤治疗效果及视力改善程度,放疗优于化疗。
儿童视路胶质瘤天坛诊疗规范(2021)
视路胶质瘤(OPG)是儿童常见鞍区肿瘤,迄今为止,国内外均没有标准的诊疗规范,治疗效果差异巨大。在此,我们提出OPG整合诊断及治疗规范,与同道共享,希望大家汲取我们的经验教训,少走弯路,提高疗效,造福广大患儿。
[1]Jahraus C D, Tarbell N J. Optic pathway gliomas[J]. Pediatr Blood Cancer, 2006,46(5):586-596.
[2]Rasool N, Odel J G, Kazim M. Optic pathway glioma of childhood[J]. Curr Opin Ophthalmol, 2017,28(3):289-295.[3]Aihara Y, Chiba K, Eguchi S, et al. Pediatric Optic Pathway/Hypothalamic Glioma[J]. Neurol Med Chir (Tokyo), 2018,58(1):1-9.[4]DODGE H J, LOVE J G, CRAIG W M, et al. Gliomas of the optic nerves[J]. AMA Arch Neurol Psychiatry, 1958,79(6):607-621.[5]Liu Y, Hao X, Liu W, et al. Analysis of Survival Prognosis for Children with Symptomatic Optic Pathway Gliomas Who Received Surgery[J]. World Neurosurg, 2018,109:e1-e15.[6]Ryall S, Tabori U, Hawkins C. Pediatric low-grade glioma in the era of molecular diagnostics[J]. Acta Neuropathol Commun, 2020,8(1):30.[7]Munoz-Cardona M L, Llano-Naranjo Y, Londono-Ocampo F, et al. Acute Visual Loss Related to Retinal Vascular Occlusion Secondary to Visual Pathway Primary Glioblastoma[J]. J Neuroophthalmol, 2021,41(2):e142-e144.[8]Lin C Y, Huang H M. Unilateral malignant optic glioma following glioblastoma multiforme in the young: a case report and literature review[J]. BMC Ophthalmol, 2017,17(1):21.[9]Siedler D G, Beechey J C, Jessup P J, et al. Infantile Optic Pathway Glioblastoma[J]. World Neurosurg, 2019,129:172-175.[10]朱惠娟. 鞍区病变的诊断与治疗[J]. 中华内科杂志, 2016,55(1):49-50.[11]白光辉, 邹爱国, 严志汉, 等. 儿童鞍区常见肿瘤26例影像学分析[J]. 浙江医学, 2006,28(4):304-306.[12]Jagannathan J, Dumont A S, Jane J J, et al. Pediatric sellar tumors: diagnostic procedures and management[J]. Neurosurg Focus, 2005,18(6A):E6.[13]Fried I, Tabori U, Tihan T, et al. Optic pathway gliomas: a review[J]. CNS Oncol, 2013,2(2):143-159.[14]Pak-Yin L A, Moreira D C, Sun C, et al. Challenges and opportunities for managing pediatric central nervous system tumors in China[J]. Pediatr Investig, 2020,4(3):211-217.[15]周大彪, 罗世祺, 马振宇, 等. 1267例儿童神经系统肿瘤的流行病学[J]. 中华神经外科杂志, 2007,23(1):4-7.[16]李莉红, 李玉华, 郑慧, 等. 儿童鞍区占位性病变的临床与影像学特征[J]. 实用放射学杂志, 2017,33(4):593-596, 652.[17]刘博, 廖宇翔, 张治平, 等. 儿童鞍区肿瘤的治疗策略[J]. 国际神经病学神经外科学杂志, 2018,45(4):383-386.[18]幸兵, 郭晓鹏, 姚勇, 等. 儿童鞍区生殖细胞肿瘤的早期诊断及治疗策略: 第三届全国小儿神经外科学术大会, 重庆, 2015[C].[19]宫剑, 甲戈, 张玉琪, 等. 鞍区生殖细胞瘤的早期诊断及综合治疗[J]. 中国微侵袭神经外科杂志, 2012,17(6):245-247.[20]Keil M F, Stratakis C A. Pituitary tumors in childhood: update of diagnosis, treatment and molecular genetics[J]. Expert Rev Neurother, 2008,8(4):563-574.[21]邢亚洲, 王新军. 儿童垂体腺瘤的临床特点及治疗[J]. 中华实用儿科临床杂志, 2017,32(18):1429-1432.[22]孙漓, 周列民, 周珏倩, 等. 中国人NF1基因突变分析[J]. 中国现代神经疾病杂志, 2007,7(4):343-346.[23]吉津, 郭琴, 章若画, 等. 1型神经纤维瘤病NF1基因突变检测[J]. 中华皮肤科杂志, 2017,50(6):442-444.[24]洪伟, 李宇杰, 贾宗良. 24例神经纤维瘤病临床分析[J]. 现代肿瘤医学, 2012,20(7):1447-1450.[25]Adil A, Singh A K. Neurofibromatosis Type 1[J]. 2021.[26]Evans D G, Moran A, King A, et al. Incidence of vestibular schwannoma and neurofibromatosis 2 in the North West of England over a 10-year period: higher incidence than previously thought[J]. Otol Neurotol, 2005,26(1):93-97.[27]Evans D G, Howard E, Giblin C, et al. Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service[J]. Am J Med Genet A, 2010,152A(2):327-332.[28]Smith M J, Higgs J E, Bowers N L, et al. Cranial meningiomas in 411 neurofibromatosis type 2 (NF2) patients with proven gene mutations: clear positional effect of mutations, but absence of female severity effect on age at onset[J]. J Med Genet, 2011,48(4):261-265.[29]Parry D M, Eldridge R, Kaiser-Kupfer M I, et al. Neurofibromatosis 2 (NF2): clinical characteristics of 63 affected individuals and clinical evidence for heterogeneity[J]. Am J Med Genet, 1994,52(4):450-461.[30]Dutton J J. Gliomas of the anterior visual pathway[J]. Survey of Ophthalmology, 1994,38(5):427-452.[31]Listernick R, Charrow J, Greenwald M, et al. Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study[J]. J Pediatr, 1994,125(1):63-66.[32]Sellmer L, Farschtschi S, Marangoni M, et al. Serial MRIs provide novel insight into natural history of optic pathway gliomas in patients with neurofibromatosis 1[J]. Orphanet J Rare Dis, 2018,13(1):62.[33]Cassina M, Frizziero L, Opocher E, et al. Optic Pathway Glioma in Type 1 Neurofibromatosis: Review of Its Pathogenesis, Diagnostic Assessment, and Treatment Recommendations[J]. Cancers (Basel), 2019,11(11).[34]Louis D N, Perry A, Reifenberger G, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary[J]. Acta Neuropathol, 2016,131(6):803-820.[35]Louis D N, Perry A, Wesseling P, et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary[J]. Neuro Oncol, 2021,23(8):1231-1251.[36]Kornreich L, Blaser S, Schwarz M, et al. Optic pathway glioma: correlation of imaging findings with the presence of neurofibromatosis[J]. AJNR Am J Neuroradiol, 2001,22(10):1963-1969.[37]Jones D T, Kocialkowski S, Liu L, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas[J]. Cancer Res, 2008,68(21):8673-8677.[38]Mistry M, Zhukova N, Merico D, et al. BRAF mutation and CDKN2A deletion define a clinically distinct subgroup of childhood secondary high-grade glioma[J]. J Clin Oncol, 2015,33(9):1015-1022.[39]Packer R J, Iavarone A, Jones D, et al. Implications of new understandings of gliomas in children and adults with NF1: report of a consensus conference[J]. Neuro Oncol, 2020,22(6):773-784.[40]Ryall S, Tabori U, Hawkins C. Pediatric low-grade glioma in the era of molecular diagnostics[J]. Acta Neuropathol Commun, 2020,8(1):30.[41]Friedrich R E, Nuding M A. Optic Pathway Glioma and Cerebral Focal Abnormal Signal Intensity in Patients with Neurofibromatosis Type 1: Characteristics, Treatment Choices and Follow-up in 134 Affected Individuals and a Brief Review of the Literature[J]. Anticancer Res, 2016,36(8):4095-4121.[42]Listernick R, Ferner R E, Piersall L, et al. Late-onset optic pathway tumors in children with neurofibromatosis 1[J]. Neurology, 2004,63(10):1944-1946.[43]Jenkin D, Angyalfi S, Becker L, et al. Optic glioma in children: surveillance, resection, or irradiation?[J]. Int J Radiat Oncol Biol Phys, 1993,25(2):215-225.[44]Dodgshun A J, Elder J E, Hansford J R, et al. Long-term visual outcome after chemotherapy for optic pathway glioma in children: Site and age are strongly predictive[J]. Cancer, 2015,121(23):4190-4196.[45]Awdeh R M, Kiehna E N, Drewry R D, et al. Visual outcomes in pediatric optic pathway glioma after conformal radiation therapy[J]. Int J Radiat Oncol Biol Phys, 2012,84(1):46-51.[46]Listernick R, Ferner R E, Liu G T, et al. Optic pathway gliomas in neurofibromatosis-1: controversies and recommendations[J]. Ann Neurol, 2007,61(3):189-198.[47]Bataini J P, Delanian S, Ponvert D. Chiasmal gliomas: results of irradiation management in 57 patients and review of literature[J]. Int J Radiat Oncol Biol Phys, 1991,21(3):615-623.[48]Binning M J, Liu J K, Kestle J R, et al. Optic pathway gliomas: a review[J]. Neurosurg Focus, 2007,23(5):E2.[49]Packer R J, Lange B, Ater J, et al. Carboplatin and vincristine for recurrent and newly diagnosed low-grade gliomas of childhood[J]. J Clin Oncol, 1993,11(5):850-856.[50]Cappelli C, Grill J, Raquin M, et al. Long-term follow up of 69 patients treated for optic pathway tumours before the chemotherapy era[J]. Arch Dis Child, 1998,79(4):334-338.[51]Hidalgo E T, Kvint S, Orillac C, et al. Long-term clinical and visual outcomes after surgical resection of pediatric pilocytic/pilomyxoid optic pathway gliomas[J]. J Neurosurg Pediatr, 2019,24(2):166-173.[52]Grabenbauer G G, Schuchardt U, Buchfelder M, et al. Radiation therapy of optico-hypothalamic gliomas (OHG)--radiographic response, vision and late toxicity[J]. Radiother Oncol, 2000,54(3):239-245.[53]Laithier V, Grill J, Le Deley M C, et al. Progression-free survival in children with optic pathway tumors: dependence on age and the quality of the response to chemotherapy--results of the first French prospective study for the French Society of Pediatric Oncology[J]. J Clin Oncol, 2003,21(24):4572-4578.[54]Massimino M, Spreafico F, Cefalo G, et al. High response rate to cisplatin/etoposide regimen in childhood low-grade glioma[J]. J Clin Oncol, 2002,20(20):4209-4216.[55]Sawamura Y, Kamada K, Kamoshima Y, et al. Role of surgery for optic pathway/hypothalamic astrocytomas in children[J]. Neuro Oncol, 2008,10(5):725-733.[56]田永吉, 李德岭, 甲戈, 等. 53例儿童视路胶质瘤的临床特点及预后分析[J]. 中华神经外科杂志, 2012,28(11):1137-1140.[57]Ullrich N J, Prabhu S P, Reddy A T, et al. A phase II study of continuous oral mTOR inhibitor everolimus for recurrent, radiographic-progressive neurofibromatosis type 1-associated pediatric low-grade glioma: a Neurofibromatosis Clinical Trials Consortium study[J]. Neuro Oncol, 2020,22(10):1527-1535.[58]Hill C S, Khan M, Phipps K, et al. Neurosurgical experience of managing optic pathway gliomas[J]. Childs Nerv Syst, 2021,37(6):1917-1929.[59]Inskip P D, Curtis R E. New malignancies following childhood cancer in the United States, 1973-2002[J]. Int J Cancer, 2007,121(10):2233-2240.[60]Morikawa M, Tamaki N, Kokunai T, et al. Cerebellar pilocytic astrocytoma with leptomeningeal dissemination: case report[J]. Surg Neurol, 1997,48(1):49-51, 51-52.[61]Pollack I F, Hurtt M, Pang D, et al. Dissemination of low grade intracranial astrocytomas in children[J]. Cancer, 1994,73(11):2869-2878.[62]Bian S X, McAleer M F, Vats T S, et al. Pilocytic astrocytoma with leptomeningeal dissemination[J]. Childs Nerv Syst, 2013,29(3):441-450.[63]Abel T J, Chowdhary A, Thapa M, et al. Spinal cord pilocytic astrocytoma with leptomeningeal dissemination to the brain. Case report and review of the literature[J]. J Neurosurg, 2006,105(6 Suppl):508-514.[64]Aryan H E, Meltzer H S, Lu D C, et al. Management of pilocytic astrocytoma with diffuse leptomeningeal spread: two cases and review of the literature[J]. Childs Nerv Syst, 2005,21(6):477-481.[65]Terasaki M, Bouffet E, Maeda M, et al. Successful treatment of leptomeningeal gliomatosis of pilomyxoid astrocytoma after failed frontline chemotherapy[J]. Neurologist, 2012,18(1):32-35.[66]Ryall S, Tabori U, Hawkins C. Pediatric low-grade glioma in the era of molecular diagnostics[J]. Acta Neuropathol Commun, 2020,8(1):30.[67]Bavle A, Jones J, Lin F Y, et al. Dramatic clinical and radiographic response to BRAF inhibition in a patient with progressive disseminated optic pathway glioma refractory to MEK inhibition[J]. Pediatr Hematol Oncol, 2017,34(4):254-259.[68]Sievert A J, Lang S S, Boucher K L, et al. Paradoxical activation and RAF inhibitor resistance of BRAF protein kinase fusions characterizing pediatric astrocytomas[J]. Proc Natl Acad Sci U S A, 2013,110(15):5957-5962.[69]Tutuka C, Andrews M C, Mariadason J M, et al. PLX8394, a new generation BRAF inhibitor, selectively inhibits BRAF in colonic adenocarcinoma cells and prevents paradoxical MAPK pathway activation[J]. Mol Cancer, 2017,16(1):112.[70]Miller C, Guillaume D, Dusenbery K, et al. Report of effective trametinib therapy in 2 children with progressive hypothalamic optic pathway pilocytic astrocytoma: documentation of volumetric response[J]. J Neurosurg Pediatr, 2017,19(3):319-324.[71]Fangusaro J, Onar-Thomas A, Young P T, et al. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: a multicentre, phase 2 trial[J]. Lancet Oncol, 2019,20(7):1011-1022.[72]Avery R A, Hwang E I, Jakacki R I, et al. Marked recovery of vision in children with optic pathway gliomas treated with bevacizumab[J]. JAMA Ophthalmol, 2014,132(1):111-114.声明:脑医汇旗下神外资讯、神介资讯、脑医咨询所发表内容之知识产权为脑医汇及主办方、原作者等相关权利人所有。未经许可,禁止进行转载、摘编、复制、裁切、录制等。经许可授权使用,亦须注明来源。欢迎转发、分享。





