



病例概况
2021年2月接诊一例来自山西朔州的10岁男性患儿(身高:150cm,体重:39kg)。主诉言语混乱伴呕吐,检查发现颅内占位14天。患儿14天前无明显诱因突发言语混乱、伴呕吐一次,持续约3小时后神志恢复正常。此后,间断出现言语混乱,劳累时头晕伴恶心,于当地医院检查发现颅内占位,遂来我院就诊。
门诊查体示:神清、言语略迟缓,自主体位,生长发育正常,余神经系统查体阴性。头颅CT显示:左侧颞顶枕叶占位,星形细胞瘤可能性大。头部MRI显示:左侧颞顶枕片状异常信号,边界不清,长T1长T2,左侧脑室轻度受压变形,无显著强化,FLAIR像呈混杂信号,DWI像无明显弥散受限,星形细胞瘤?(图1)。MRS显示:病灶感兴趣区Cho/Cr为2.088,NAA/Cr为0.7;正常对照区域Cho/Cr为0.805,NAA/Cr为2.0。Cho峰升高,NAA峰下降,提示低级别胶质瘤。24小时视频脑电显示为儿童正常脑电图。
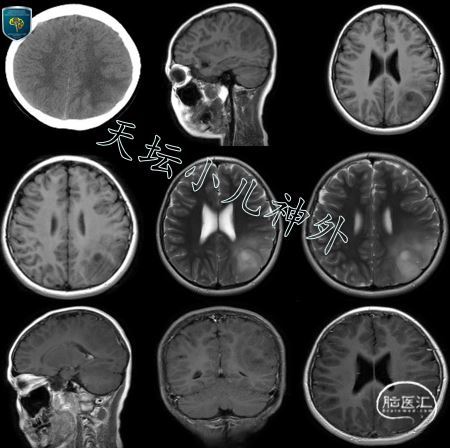
图1 术前头颅CT:左侧颞顶枕叶占位,星形细胞瘤可能性大;头部MRI显示:左侧颞顶枕片状异常信号,边界不清,长T1长T2,左侧脑室轻度受压变形,无显著强化,FLAIR像呈混杂信号,DWI像无明显弥散受限,星形细胞瘤?

患儿左侧顶枕叶占位明确,鉴于癫痫起病,影像显示占位征、瘤周水肿均不明显,无显著强化,首先考虑DNT等低级别神经上皮性肿瘤,手术指征明确。完善术前检查,于2021年2月19日在全麻下行导航辅助左顶枕开颅肿瘤切除术。术中导航及超声辅助,准确划定病变范围,电生理功能监测避开皮层运动区,见肿瘤位于中央后回后方,色灰、质韧、血供中等,大小约3.0x3.5x4.0cm,边界欠清晰。扩大切除范围,沿胶质增生带全切肿瘤,左侧脑室枕角未开放。术中超声再次探查未见肿瘤残留(图2)。手术顺利,术中出血约100ml,未输血。术毕安返ICU监护。
![]()
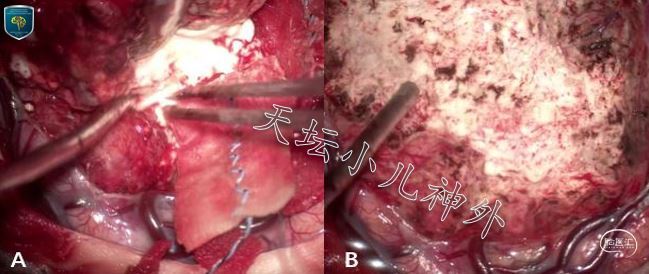
图2 术中所见:A、术中导航及超声定位,肿瘤位于中央后回后方,色灰、质韧、血供中等,边界欠清晰。B、肿瘤扩大切除,超声探查,未见残留。
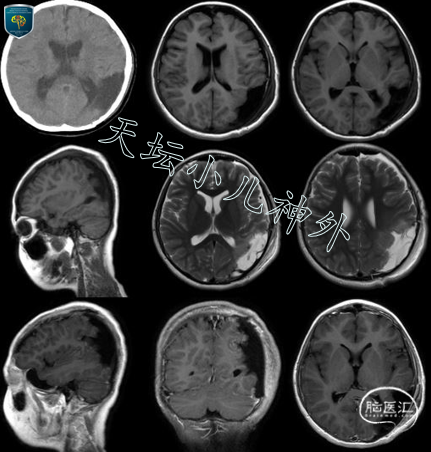
图3 术后复查头颅CT/MRI显示肿瘤切除满意。
![]()
术后患儿状态好,当晚及术后一周复查头颅CT/MRI显示肿瘤切除满意。病理回报示:间变性星形细胞瘤(WHOⅢ级),肿瘤累及软膜下。免疫组化显示:GFAP(+),Olig-2(+),P53(+),ATRX(偶见+),IDH1(+),Ki-67(约10-30%),MGMT(-)。基因检测显示:IDH1、NF1、ATRX、PTEN、KRAS突变,无1p/19q双缺失。依据第五版(2021)WHOCNS肿瘤分类,整合诊断为儿童型弥漫性高级别胶质瘤,NEC型(CNSWHO 3级)。术后患儿神清、言语同前,自主体位,未见新增神经系统阳性体征,KPS评分90分,术后两周顺利出院,继续后续标准治疗。
![]()
治疗体会
如前所述,本例首先应考虑DNT或其它低级别胶质瘤。术后病理证实间变性星形细胞瘤(WHOⅢ级,Ki-67:10-30%),确实出人意料。复习文献,间变性星形细胞瘤MRI典型表现为长T1长T2,结节样显著强化,瘤周血管源性脑水肿明显,本例显然不符。但有报道,大约三分之一的间变性星形细胞瘤无明显强化,且不伴瘤周水肿[1]。探讨原因,近四分之一的间变性星型细胞瘤是颅内原发,四分之三则由低级别胶质瘤恶性转化而来[1],本例应该属于后者。

间变性星形细胞瘤(Anaplasticastrocytoma,AA)是弥漫浸润性星形细胞肿瘤[2,3],占颅内恶性肿瘤的4%,脑胶质瘤的10%[4]。患者中位生存期为3年,5年生存率为28%[5]。AA常见于30-50岁成人,儿童少见,5年生存率低于20%,与成人无显著差异。手术切除范围是影响预后的首要因素,当肿瘤无法进行手术干预时,存活率显著下降[6]。3岁以上儿童在全切肿瘤后,尽早施行放/化疗显著延长生存期[6,7]。
基因检测已在临床中常规应用,在分子诊断中,虽然TP53和ATRX的突变最常见(>70%),但针对AA没有特异性[8]。异柠檬酸脱氢酶(IDH1/2)的突变在多数AA和继发性胶质母细胞瘤的发病机制中起着关键作用。有文献报道,间变性星形细胞瘤大致可以分为四个亚组:IDH基因突变不伴1p/19q双缺失、IDH基因突变合并1p/19q双缺失、野生型IDH不伴1p/19q双缺失、野生型IDH合并1p/19q双缺失,其中IDH基因突变合并1p/19q双缺失预后最好;IDH基因突变不伴1p/19q双缺失预后中等;野生型IDH预后最差,很多分子特征与胶质母细胞瘤相似[3]。本例IDH突变、ATRX突变,不伴1p/19q双缺失,预后中等。
有研究表明,PCV化疗(丙卡嗪、洛莫司汀、长春新碱联合)或者替莫唑胺(TMZ)单独应用与放疗的疗效未见明显差异[9]。肿瘤对治疗的敏感性与肿瘤的分子背景有关,与组织学分级无关[3]。欧洲神经肿瘤协会建议,对于缺乏1p/19q双缺失的AA,尽量全切除肿瘤并辅以单独放疗或单独烷化剂化疗(TMZ或PCV)[10]。
本例肿瘤中检测出NF1基因突变,并不能诊断为神经纤维瘤病I型;只有神经纤维瘤、视路胶质瘤中检测出NF1基因突变,才应高度怀疑,结合临床特征及染色体检查方可确诊。神经纤维瘤病含NF1型、NF2型和神经鞘瘤病[11],NF1是三种疾病中最常见的,全球每3000人中就有1人患病[12]。作为肿瘤易感综合征,患有NF1的儿童,易发生脑肿瘤,如视路胶质瘤(OPGs)、脑干胶质瘤(DIPG)[13]。NF1患者合并高级别胶质瘤的概率是普通人群的10到50倍[14]。分子机制上,神经纤维蛋白通常会抑制RAS介导的细胞生长[15],它在NF1肿瘤细胞中的丢失会导致RAS活性升高,细胞增殖和存活增加。自20世纪90年代发现NF1和NF2基因以来,相关研究不断深入,期待靶向药物的尽早开发,挽救更多的NF1、NF2患儿。
参考文献
